Predict pKa Values and Protonation States for Dynamical Systems
Protein-inhibitor binding
Henderson JA, Harris RC, Tsai CC, and Shen J*
2018How ligand protonation proton state controls water in protein ligand binding.
J Phys Chem Lett 9: 5440-5444, 2018
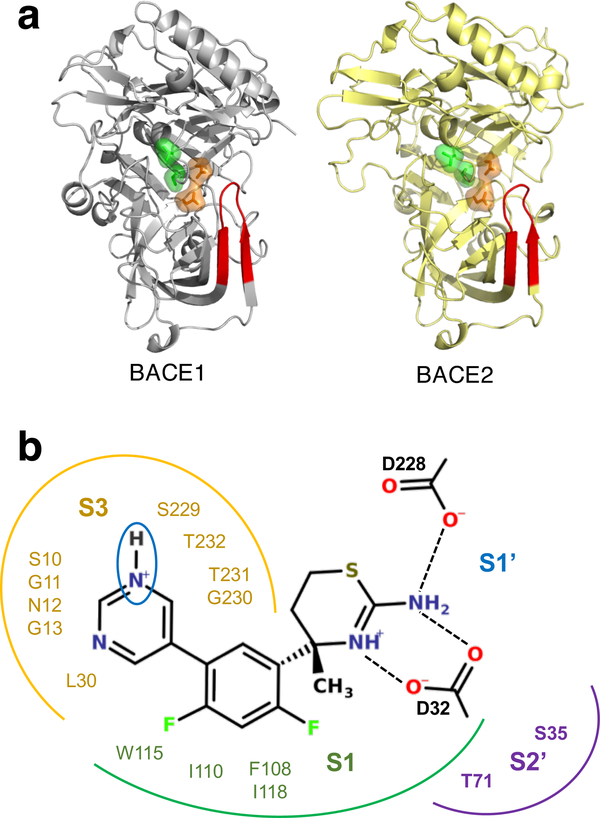
The role of water in protein-ligand binding has been an intensely studied topic in recent years; however, how ligand protonation state change perturbs water has not been considered. Here we show that water dynamics and interactions can be controlled by the protonation state of ligand using continuous constant pH molecular dynamics simulations of two closely related model systems, β-secretase 1 and 2 (BACE1 and BACE2), in complex with a small-molecule inhibitor. Simulations revealed that, upon binding, the inhibitor pyrimidine ring remains deprotonated in BACE1 but becomes protonated in BACE2. Pyrimidine protonation results in water displacement, rigidification of the binding pocket, and shift in the ligand binding mode from water-mediated to direct hydrogen bonding. These findings not only support but also rationalize the most recent structure-selectivity data in BACE1 drug design. Binding-induced protonation state changes are likely common; our work offers a glimpse at how modeling protein-ligand binding while allowing ligand titration can further advance the understanding of water and structure-based drug design.
Harris RC, Tsai CC, Ellis CR, and Shen J*
2017Proton-coupled conformational allostery modulates the inhibitor selectivity for β-secretase.
J Phys Chem Lett 8: 4832-4837, 2017
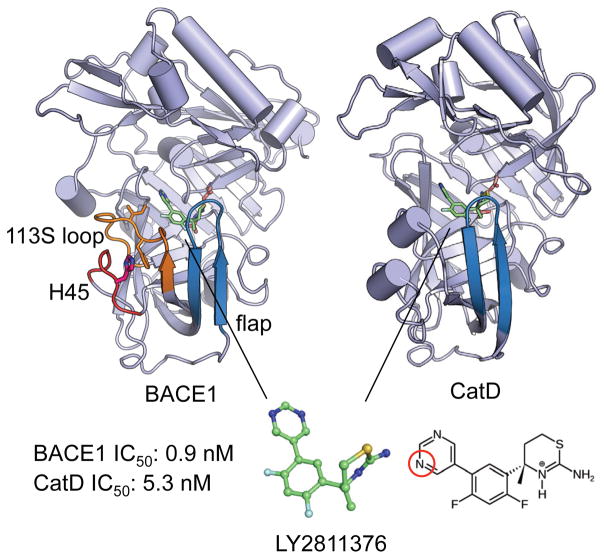
Many important pharmaceutical targets, such as aspartyl proteases and kinases, exhibit pH-dependent dynamics, functions and inhibition. Accurate prediction of their binding free energies is challenging because current computational techniques neglect the effects of pH. Here we combine free energy perturbation calculations with continuous constant pH molecular dynamics to explore the selectivity of a small-molecule inhibitor for β-secretase (BACE1), an important drug target for Alzheimer's disease. The calculations predicted identical affinity for BACE1 and the closely related cathepsin D at high pH; however, at pH 4.6 the inhibitor is selective for BACE1 by 1.3 kcal/mol, in excellent agreement with experiment.
Ellis CR, Tsai CC, Lin FY, and Shen J*
2017Conformational dynamics of cathepsin D and binding to a small-molecule BACE1 inhibitor.
J Comput Chem 38:1260-1269, 2017
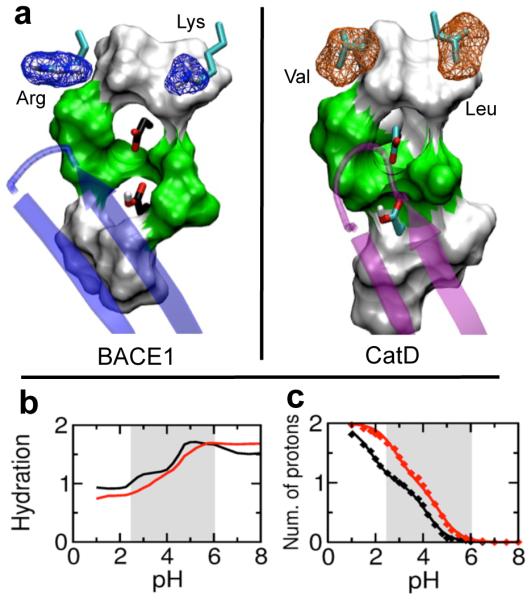
BACE1 is a major therapeutic target for prevention and treatment of Alzheimer's disease. Developing inhibitors that can selectively target BACE1 in favor of other proteases, especially cathepsin D (CatD), has presented significant challenges. Here, we investigate the conformational dynamics and protonation states of BACE1 and CatD using continuous constant pH molecular dynamics with pH replica-exchange sampling protocol. Despite similar structure, BACE1 and CatD exhibit markedly different active site dynamics. BACE1 displays pH-dependent flap dynamics that controls substrate accessibility, while the CatD flap is relatively rigid and remains open in the pH range 2.5-6. Interestingly, although each protease hydrolyzes peptide bonds, the protonation states of the catalytic dyads are different within the active pH range.
Ellis CR, Tsai CC, Hou XJ, and Shen J*
2016Constant pH molecular dynamics reveals pH-modulated binding of two small-molecule BACE1 inhibitors.
J Phys Chem Lett 7: 944-949, 2016
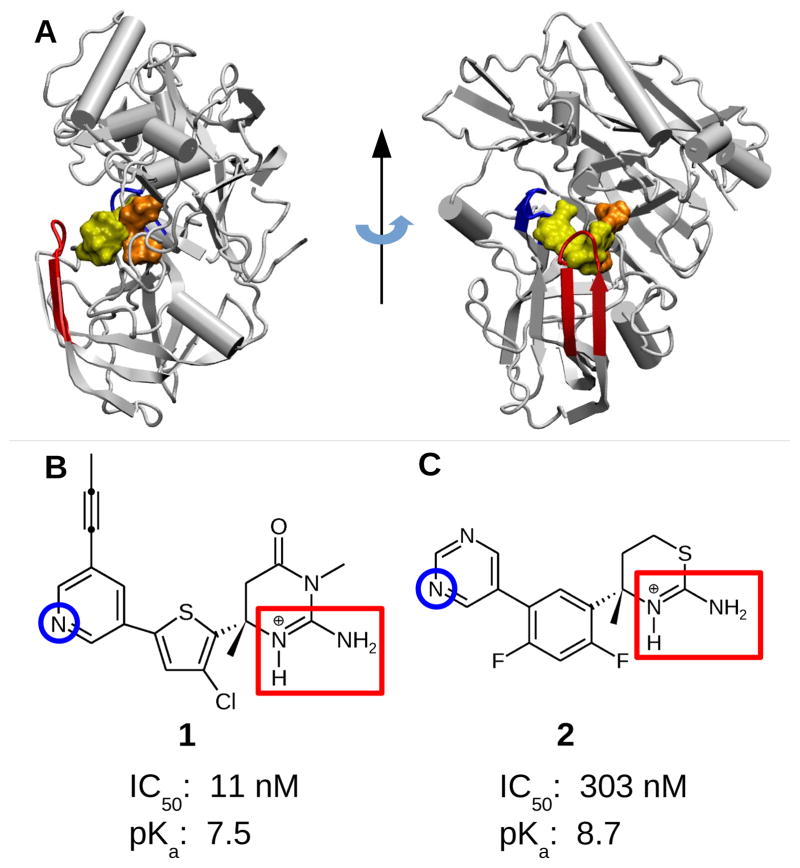
Targeting β-secretase (BACE1) with small-molecule inhibitors offers a promising route for treatment of Alzheimer's disease. However, the intricate pH dependence of BACE1 function and inhibitor efficacy has posed major challenges for structure-based drug design. Here we investigate two structurally similar BACE1 inhibitors that have dramatically different inhibitory activity using continuous constant pH molecular dynamics (CpHMD). At high pH, both inhibitors are stably bound to BACE1; however, within the enzyme active pH range, only the iminopyrimidinone-based inhibitor remains bound, while the aminothiazine-based inhibitor becomes partially dissociated following the loss of hydrogen bonding with the active site and change of the 10s loop conformation. The drastically lower activity of the second inhibitor is due to the protonation of a catalytic aspartate and the lack of a propyne tail. This work demonstrates that CpHMD can be used for screening pH-dependent binding profiles of small-molecule inhibitors, providing a new tool for structure-based drug design and optimization.
Protein-protein binding
Wallace JA and Shen JK*
2012Unraveling a trap-and-trigger mechanism in the pH-sensitive self-assembly of spider silk proteins.
J Phys Chem Lett 3: 658-662, 2012
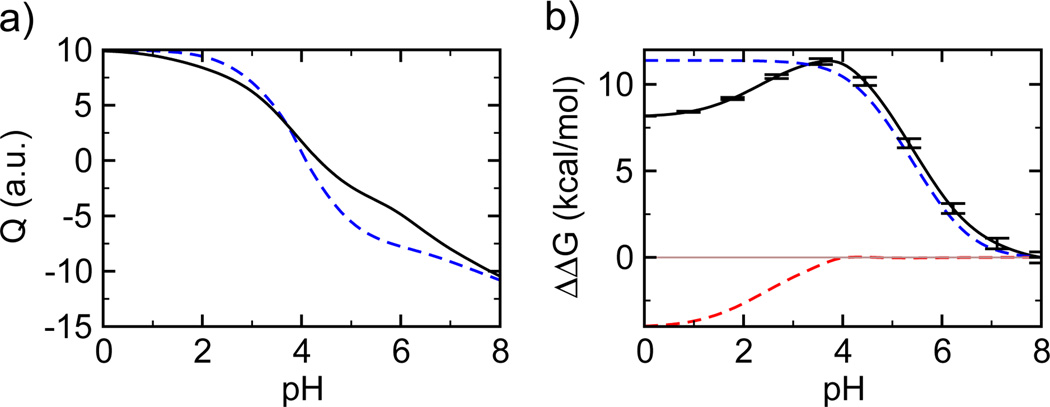
When the major ampulate spidroins (MaSp1) are called upon to form spider dragline silk, one of nature's most amazing materials, a small drop in pH must occur. Using a state-of-the-art simulation technique, constant pH molecular dynamics, we discovered a few residues that respond to the pH signal in the dimerization of the N-terminal domain (NTD) of MaSp1 which is an integral step in the fiber assembly. At neutral pH the deprotonation of Glu79 and Glu119 leads to water penetration and structural changes at the monomer-monomer binding interface. At strongly acidic pH, the protonation of Asp39 and Asp40 weakens the electrostatic attraction between the monomers. Thus, we propose a "trap-and-trigger" mechanism whereby the intermolecular salt-bridges at physiologically relevant pH conditions always act as a stabilizing "trap" favoring dimerization. As pH is lowered to about 6, Glu79 and Glu119 become protonated, triggering the dimerization and subsequent silk formation. We speculate that this type of mechanism is operative in many other pH-sensitive biological processes.
Transmembrane proteins
Henderson JA, Beckstein O, and Shen J*
2020Alternative proton-binding site and long-distance coupling in Escherichia coli sodium–proton antiporter NhaA.
Under review, 2020
Yue Z, Chen W, Zgurskaya, H, and Shen J*
2017Constant pH molecular dynamics reveals how proton release drives the conformational transition of a transmembrane efflux pump.
J Chem Theory Comput 13: 6405-6414, 2017
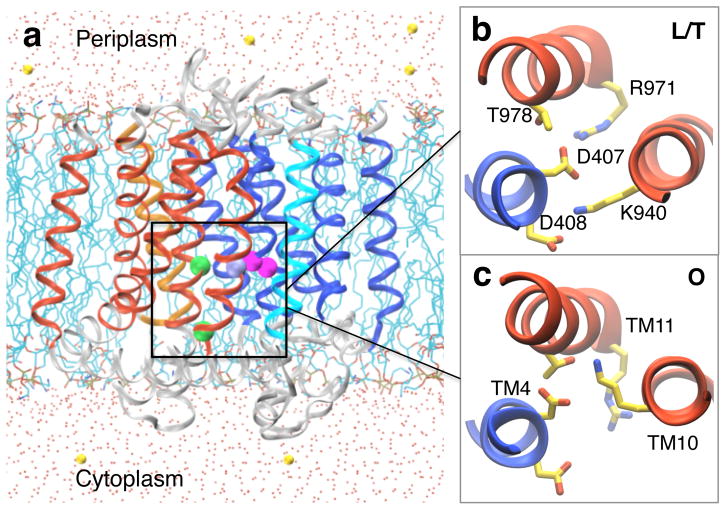
AcrB is the inner-membrane transporter of an E. coli AcrAB-TolC tripartite efflux complex, which plays a major role in the intrinsic resistance to clinically important antibiotics. AcrB pumps a wide range of toxic substrates by utilizing the proton gradient between periplasm and cytoplasm. Crystal structures of AcrB revealed three distinct conformational states of the transport cycle, substrate access, binding, and extrusion or loose (L), tight (T), and open (O) states. However, the specific residue(s) responsible for proton binding/release and the mechanism of proton-coupled conformational cycling remain controversial. Here we use the newly developed membrane hybrid-solvent continuous constant pH molecular dynamics technique to explore the protonation states and conformational dynamics of the transmembrane domain of AcrB. Simulations show that both Asp407 and Asp408 are deprotonated in the L/T states, while only Asp408 is protonated in the O state. Remarkably, release of a proton from Asp408 in the O state results in large conformational changes, such as the lateral and vertical movement of transmembrane helices as well as the salt-bridge formation between Asp408 and Lys940 and other side chain rearrangements among essential residues.
Chen W, Huang YD and Shen J*
2016Conformational activation of a transmembrane proton channel from constant pH molecular dynamics.
J Phys Chem Lett 7: 3961-3966, 2016
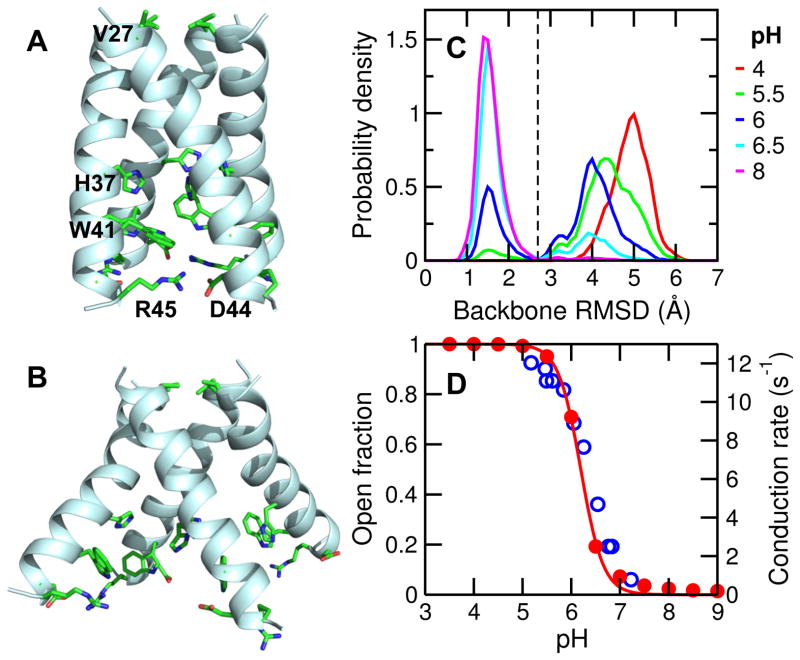
Proton-coupled transmembrane proteins play important roles in human health and diseases; however, detailed mechanisms are often elusive. Experimentally resolving proton positions and structural details is challenging, and conventional molecular dynamics simulations are performed with preassigned and fixed protonation states. To address this challenge, here we illustrate the use of the state-of-the-art continuous constant pH molecular dynamics (CpHMD) to directly describe the activation of the M2 channel of influenza virus, for which abundant experimental data are available. Starting from the closed crystal structure, simulation reveals a pH-dependent conformational switch to an activated state that resembles the open crystal structure. Importantly, simulation affords the free energy of channel opening coupled to the titration of a histidine tetrad, thereby providing a thermodynamic mechanism for M2 activation, that is consistent with NMR data and resolves the controversy with crystal structures obtained at different pH values. This work illustrates the utility of CpHMD in offering previously unattainable conformational details and thermodynamic information for proton-coupled transmembrane channels and transporters.
Huang YD, Chen W, Dotson DL, Beckstein O, and Shen J*
2016Mechanism of pH-dependent activation of the sodium-proton antiporter NhaA.
Nature Commun 7:12940, 2016
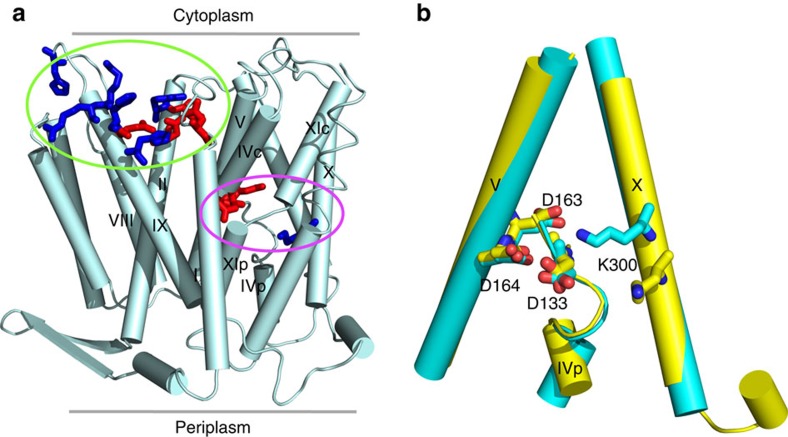
Escherichia coli NhaA is a prototype sodium-proton antiporter, which has been extensively characterized by X-ray crystallography, biochemical and biophysical experiments. However, the identities of proton carriers and details of pH-regulated mechanism remain controversial. Here we report constant pH molecular dynamics data, which reveal that NhaA activation involves a net charge switch of a pH sensor at the entrance of the cytoplasmic funnel and opening of a hydrophobic gate at the end of the funnel. The latter is triggered by charging of Asp164, the first proton carrier. The second proton carrier Lys300 forms a salt bridge with Asp163 in the inactive state, and releases a proton when a sodium ion binds Asp163. These data reconcile current models and illustrate the power of state-of-the-art molecular dynamics simulations in providing atomic details of proton-coupled transport across membrane which is challenging to elucidate by experimental techniques.
Soluble proteins and peptides
Harris RC, Liu R, and Shen J*
2020Predicting reactive cysteines with implicit-solvent based continuous constant pH molecular dynamics in Amber.
J Chem Theory Comput 16: 3689-3698, 2020
Cysteines existing in the deprotonated thiolate form or having a tendency to become deprotonated are important players in enzymatic and cellular redox functions and frequently exploited in covalent drug design; however, most computational studies assume cysteines as protonated. Thus, developing an efficient tool that can make accurate and reliable predictions of cysteine protonation states is timely needed. We recently implemented a generalized Born (GB) based continuous constant pH molecular dynamics (CpHMD) method in Amber for protein pKa calculations on CPUs and GPUs.
Huang YD, Yue Z, Tsai CC, Henderson JA, and Shen J*
2018Predicting catalytic proton donors and nucleophiles in enzymes: how adding dynamics helps elucidate the structure-function relationships.
J Phys Chem Lett 9: 1179-1184, 2018
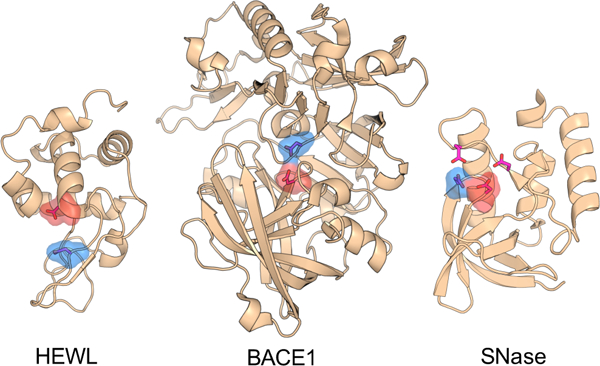
Despite the relevance of understanding structure-function relationships, robust prediction of proton donors and nucleophiles in enzyme active sites remains challenging. Here we tested three types of state-of-the-art computational methods to calculate the p Ka's of the buried and hydrogen bonded catalytic dyads in five enzymes. We asked the question what determines the p Ka order, i.e., what makes a residue proton donor vs a nucleophile. The continuous constant pH molecular dynamics simulations captured the experimental p Ka orders and revealed that the negative nucleophile is stabilized by increased hydrogen bonding and solvent exposure as compared to the proton donor. Surprisingly, this simple trend is not apparent from crystal structures and the static structure-based calculations. While the generality of the findings awaits further testing via a larger set of data, they underscore the role of dynamics in bridging enzyme structures and functions.
Huang YD, Chen W, and Shen J*
2016Continuous constant pH molecular dynamics with particle-mesh Ewald and titratabe water.
J Chem Theory Comput. 12: 5411-5421, 2016
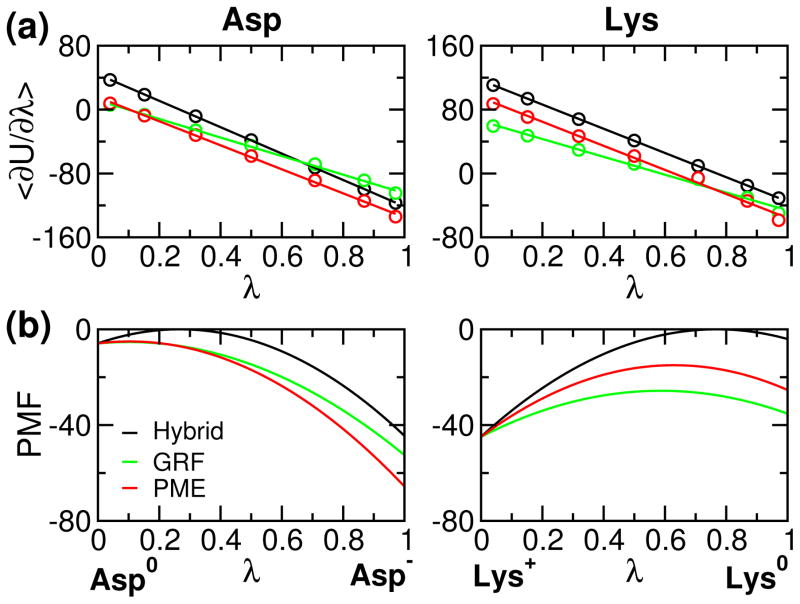
Development of a pH stat to properly control solution pH in biomolecular simulations has been a long-standing goal in the community. Toward this goal recent years have witnessed the emergence of the so-called constant pH molecular dynamics methods. However, the accuracy and generality of these methods have been hampered by the use of implicit-solvent models or truncation-based electrostatic schemes. Here we report the implementation of the particle mesh Ewald (PME) scheme into the all-atom continuous constant pH molecular dynamics (CpHMD) method, enabling CpHMD to be performed with a standard MD engine at a fractional added computational cost. We demonstrate the performance using pH replica-exchange CpHMD simulations with titratable water for a stringent test set of proteins, HP36, BBL, HEWL, and SNase.
Ellis CR and Shen J*
2015pH-dependent population shift regulates BACE1 activity and inhibition.
J Am Chem Soc 137: 9543-9546, 2015
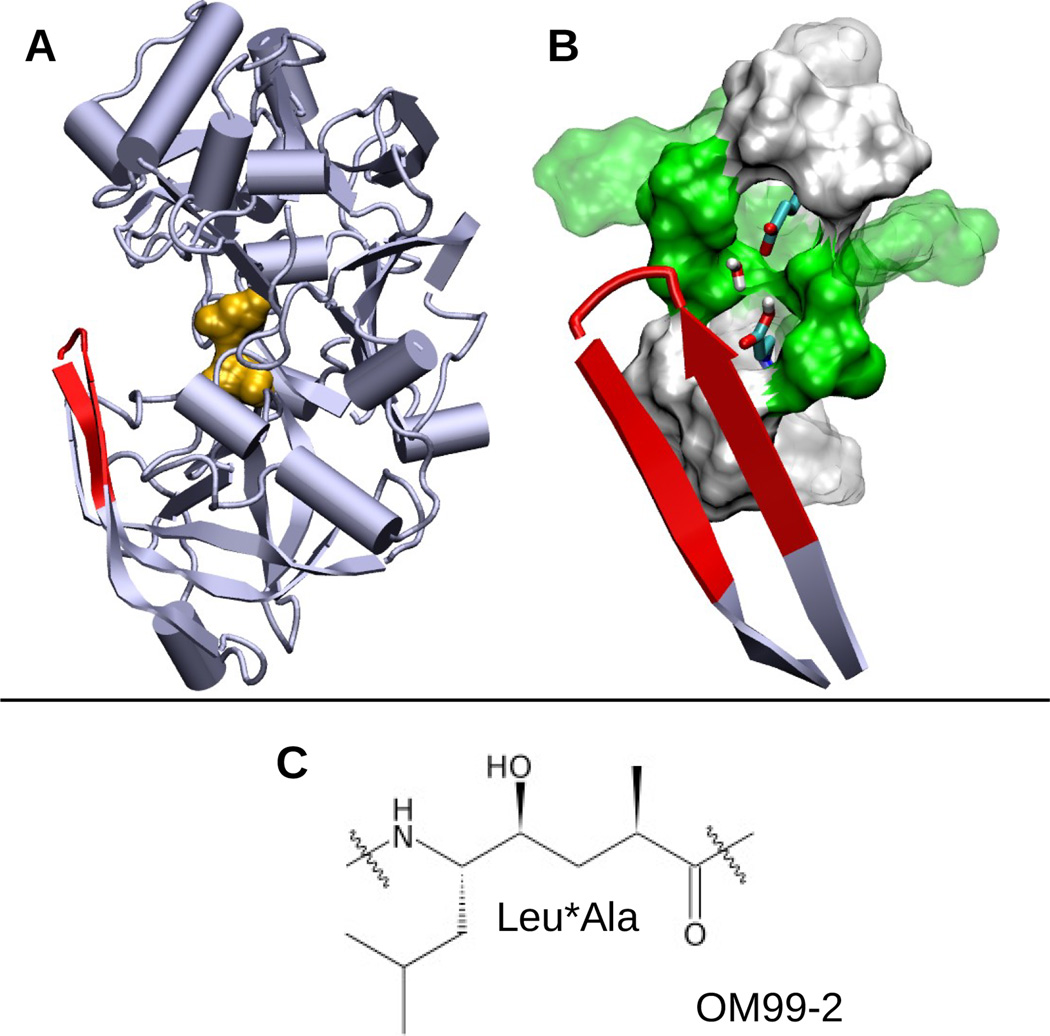
BACE1, a major therapeutic target for treatment of Alzheimer's disease, functions within a narrow pH range. Despite tremendous effort and progress in the development of BACE1 inhibitors, details of the underlying pH-dependent regulatory mechanism remain unclear. Here we elucidate the pH-dependent conformational mechanism that regulates BACE1 activity using continuous constant-pH molecular dynamics (MD). The simulations reveal that BACE1 mainly occupies three conformational states and that the relative populations of the states shift according to pH. At intermediate pH, when the catalytic dyad is monoprotonated, a binding-competent state is highly populated, while at low and high pH a Tyr-inhibited state is dominant. Our data provide strong evidence supporting conformational selection as a major mechanism for substrate and peptide-inhibitor binding. These new insights, while consistent with experiment, greatly extend the knowledge of BACE1 and have implications for further optimization of inhibitors and understanding potential side effects of targeting BACE1. Finally, the work highlights the importance of properly modeling protonation states in MD simulations.
Shi C, Wallace JA, and Shen JK*
2012Thermodynamic coupling of protonation and conformational equilibria in proteins: theory and simulation.
Biophys J. 102: 1590-1597, 2012
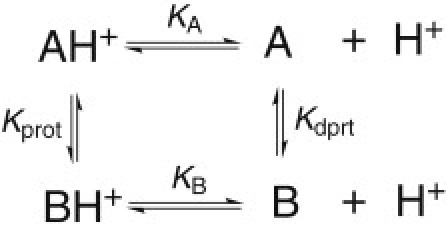
Ionization-coupled conformational phenomena are ubiquitous in biology. However, quantitative characterization of the underlying thermodynamic cycle comprised of protonation and conformational equilibria has remained an elusive goal. Here we use theory and continuous constant pH molecular dynamics (CpHMD) simulations to provide a thermodynamic description for the coupling of proton titration and conformational exchange between two distinct states of a protein. CpHMD simulations with a hybrid-solvent scheme and the pH-based replica-exchange (REX) protocol are applied to obtain the equilibrium constants and atomic details of the ionization-coupled conformational exchange between open and closed states of an engineered mutant of staphylococcal nuclease. Although the coupling of protonation and conformational equilibria is not exact in the simulation, the results are encouraging. They demonstrate that REX-CpHMD simulations can be used to study thermodynamics of ionization-coupled conformational processes--which has not possible using present experimental techniques or traditional simulations based on fixed protonation states.
Wallace JA and Shen JK*
2011Continuous constant pH molecular dynamics in explicit solvent with pH-based replica exchange.
J Chem Theory Comput 7: 2617-2629, 2011
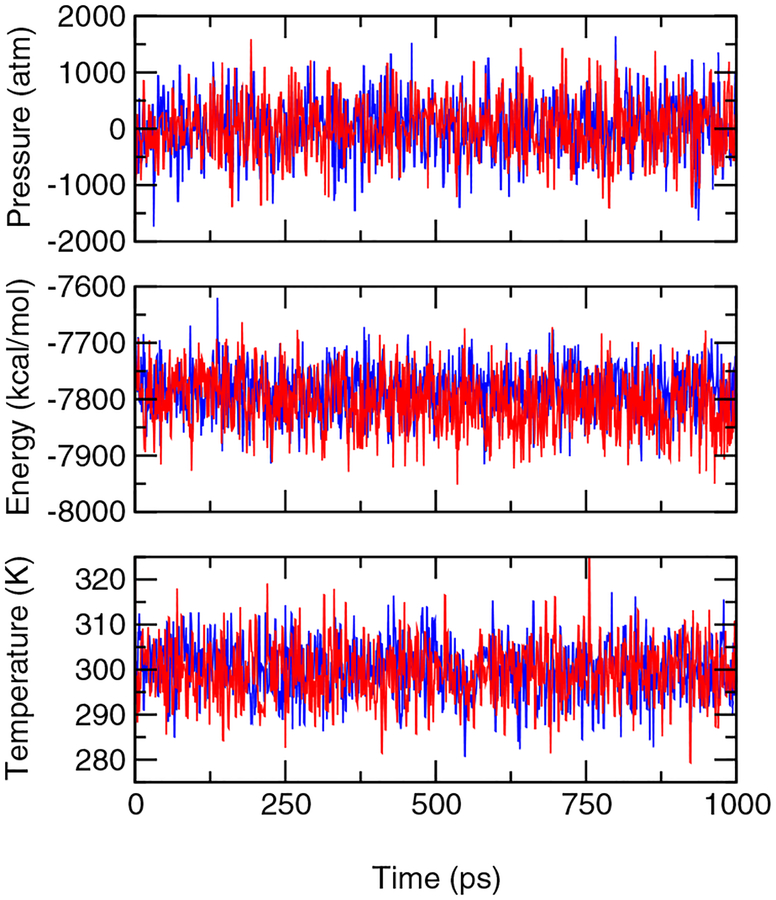
A computational tool that offers accurate pKa values and atomically detailed knowledge of protonation-coupled conformational dynamics is valuable for elucidating mechanisms of energy transduction processes in biology, such as enzyme catalysis and electron transfer as well as proton and drug transport. Toward this goal we present a new technique of embedding continuous constant pH molecular dynamics within an explicit-solvent representation. In this technique we make use of the efficiency of the generalized-Born (GB) implicit-solvent model for estimating the free energy of protein solvation while propagating conformational dynamics using the more accurate explicit-solvent model. Also, we employ a pH-based replica exchange scheme to significantly enhance both protonation and conformational state sampling. Benchmark data of five proteins including HP36, NTL9, BBL, HEWL, and SNase yield an average absolute deviation of 0.53 and a root mean squared deviation of 0.74 from experimental data. This level of accuracy is obtained with 1 ns simulations per replica. Detailed analysis reveals that explicit-solvent sampling provides increased accuracy relative to the previous GB-based method by preserving the native structure, providing a more realistic description of conformational flexibility of the hydrophobic cluster, and correctly modeling solvent mediated ion-pair interactions. Thus, we anticipate that the new technique will emerge as a practical tool to capture ionization equilibria while enabling an intimate view of ionization coupled conformational dynamics that is difficult to delineate with experimental techniques alone.
Wallace JA, Wang Y, Shi C, Pastoor KJ, Nguyen B-L, Xia K, and Shen JK*
2011Towards accurate prediction of pKa values for protein internal residues: the importance of conformational relaxation and desolvation energy.
Proteins 79: 3364-3363, 2011
Proton uptake or release controls many important biological processes, such as energy transduction, virus replication, and catalysis. Accurate pK(a) prediction informs about proton pathways, thereby revealing detailed acid-base mechanisms. Physics-based methods in the framework of molecular dynamics simulations not only offer pK(a) predictions but also inform about the physical origins of pK(a) shifts and provide details of ionization-induced conformational relaxation and large-scale transitions. One such method is the recently developed continuous constant pH molecular dynamics (CPHMD) method, which has been shown to be an accurate and robust pK(a) prediction tool for naturally occurring titratable residues. To further examine the accuracy and limitations of CPHMD, we blindly predicted the pK(a) values for 87 titratable residues introduced in various hydrophobic regions of staphylococcal nuclease and variants. The predictions gave a root-mean-square deviation of 1.69 pK units from experiment, and there were only two pK(a)'s with errors greater than 3.5 pK units. Analysis of the conformational fluctuation of titrating side-chains in the context of the errors of calculated pK(a) values indicate that explicit treatment of conformational flexibility and the associated dielectric relaxation gives CPHMD a distinct advantage. Analysis of the sources of errors suggests that more accurate pK(a) predictions can be obtained for the most deeply buried residues by improving the accuracy in calculating desolvation energies. Furthermore, it is found that the generalized Born implicit-solvent model underlying the current CPHMD implementation slightly distorts the local conformational environment such that the inclusion of an explicit-solvent representation may offer improvement of accuracy.
Wallace JA and Shen JK*
2009Predicting protein pKa’s with continuous constant pH molecular dynamics.
Methods Enzymol 66: 455-475, 2009
Knowledge of pK(a) values is important for understanding structure and function relationships in proteins. Over the past two decades, theoretical methods for pK(a) calculations have been mainly based on macroscopic models, in which the protein is considered as a low-dielectric cavity embedded in a high-dielectric continuum. In recent years, constant pH molecular dynamics methods have been developed based on a microscopic description of the protein. We describe here the methodology of continuous constant pH molecular dynamics (CPHMD), which has emerged as one of the most robust and accurate tools for predicting protein pK(a)s and for the study of pH-modulated conformational dynamics. We illustrate the utility of CPHMD by the calculation of pK(a)s for surface residues in ribonuclease A, buried residues in staphylococcal nuclease, and titratable groups in the intrinsically flexible protein α-lactalbumin. We will compare the CPHMD results with experimental data as well as calculations from PB-based and empirical methods. These examples demonstrate the accuracy and robustness of the CPHMD method and its ability to capture the correlation between ionization equilibria and conformational dynamics as well as the local dielectric response to structural rearrangement. Finally, we discuss future improvement of the CPHMD method.
Qian J, Khandogin J, West AH, Cook P*
2009Evidence for a catalytic dyad in the active site of homocitrate synthase from saccaromyces cerevisiae.
Biochemistry 47: 6851-6858, 2009
Homocitrate synthase (acetyl-coenzyme A: 2-ketoglutarate C-transferase; E.C. 2.3.3.14) (HCS) catalyzes the condensation of acetyl-CoA (AcCoA) and alpha-ketoglutarate (alpha-KG) to give homocitrate and CoA. Although the structure of an HCS has not been solved, the structure of isopropylmalate synthase (IPMS), a homologue, has been solved (Koon, N., Squire, C. J., and Baker, E. N. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 8295-8300). Three active site residues in IPMS, Glu-218, His-379, and Tyr-410, were proposed as candidates for catalytic residues involved in deprotonation of the methyl group of AcCoA prior to the Claisen condensation to give homocitrylCoA. All three of the active site residues in IPMS are conserved in the HCS from Saccharomyces cerevisiae. Site-directed mutagenesis has been carried out to probe the role of the homologous residues, Glu-155, His-309, and Tyr-320, in the S. cerevisiae HCS.....
Khandogin J and Brooks CL III*
2007Linking folding with aggregation in Alzheimer’s beta-amyloid peptides.
Proc Natl Acad Sci USA 104: 16880-16885, 2007
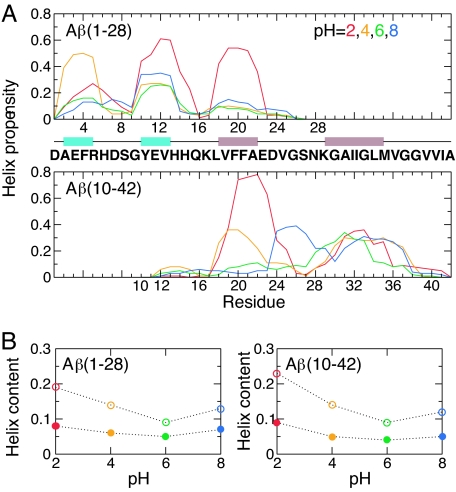
Growing evidence suggests that the beta-amyloid (Abeta) peptides of Alzheimer's disease are generated in early endosomes and that small oligomers are the principal toxic species. We sought to understand whether and how the solution pH, which is more acidic in endosomes than the extracellular environment, affects the conformational processes of Abeta. Using constant pH molecular dynamics simulations of two model peptides, Abeta(1-28) and Abeta(10-42), we found that the folding landscape of Abeta is strongly modulated by pH and is most favorable for hydrophobically driven aggregation at pH 6. Thus, our theoretical findings substantiate the possibility that Abeta oligomers develop intracellularly before secretion into the extracellular milieu, where they may disrupt synaptic activity or act as seeds for plaque formation.
Khandogin J, Raleigh DP, and Brooks CL III*
2007Folding intermediate in the villin headpiece domain arises from disruption of an N-terminal hydrogen-bonded network.
J Am Chem Soc 129: 3056-3057, 2007
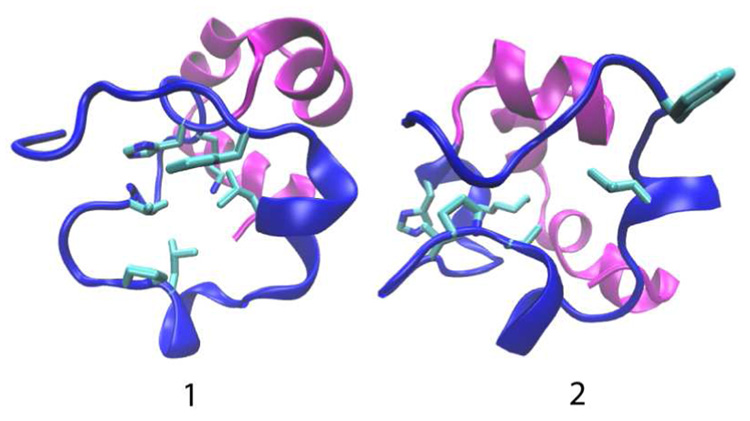
The critical role of partially folded intermediates in protein misfolding and amyloid formation has sparked interest in exploring factors that control the formation of these meta-stable species. Recent NMR experiments reported a sparsely populated intermediate of the villin headpiece domain, in which the N-terminal subdomain is random coil like under native conditions. Here we use continuous constant pH molecular dynamics simulations with replica-exchange sampling protocol to test the hypothesis that the conformational state obtained by simulations derived from a solution NMR structure represents a putative intermediate in which the N-terminal subdomain is partially unfolded. Based on the unique titration behavior of His41 as well as the structural and dynamic properties of this state, we propose that the loss of a hydrogen bond between Nδ of His41 and the backbone carbonyl oxygen of E14 in the NMR structure leads to the loss of inter-subdomain contacts as well as partial disruption of the N-terminal hydrophobic cluster. Thus, the loss of the hydrogen-bonded network and not the protonation of His41 is a prerequisite for unfolding.
Khandogin J, Chen J, and Brooks CL III*
2006Exploring atomistic details of pH-dependent peptide folding.
Proc Natl Acad Sci USA 103: 18546-18550, 2006
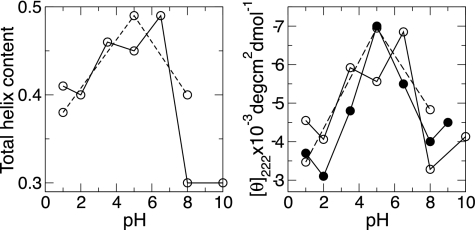
Modeling pH-coupled conformational dynamics allows one to probe many important pH-dependent biological processes, ranging from ATP synthesis, enzyme catalysis, and membrane fusion to protein folding/misfolding and amyloid formation. This work illustrates the strengths and capabilities of continuous constant pH molecular dynamics in exploring pH-dependent conformational transitions in proteins by revisiting an experimentally well studied model protein fragment, the C peptide from ribonuclease A. The simulation data reveal a bell-shaped pH profile for the total helix content, in agreement with experiment, and several pairs of electrostatic interactions that control the relative populations of unfolded and partially folded states of various helical lengths. The latter information greatly complements and extends that attainable by current experimental techniques. The present work paves the way for new and exciting applications, such as the study of pH-dependent molecular mechanism in the formation of amyloid comprising peptides from Alzheimer's and Parkinson's diseases.
Polysaccharides
Tsai CC, Payne GF, and Shen J*
2018Exploring pH-responsive, switchable crosslinking mechanisms for programming reconfigurable hydrogels based on aminopolysaccharides.
Chem Mater 30: 8597-8605, 2018
Yan K, Liu Y, Zhang J, Correa SO, Shang W, Tsai CC, Bentley WE, Shen J, Scarcelli G, Raub CB, Shi XW, Payne GF
2018Electrical programming of soft matter: using temporally varying electrical inputs to spatially control self assembly.
Biomacromolecules 19: 364-373, 2018
The growing importance of hydrogels in translational medicine has stimulated the development of top-down fabrication methods, yet often these methods lack the capabilities to generate the complex matrix architectures observed in biology. Here we show that temporally varying electrical signals can cue a self-assembling polysaccharide to controllably form a hydrogel with complex internal patterns. Evidence from theory and experiment indicate that internal structure emerges through a subtle interplay between the electrical current that triggers self-assembly and the electrical potential (or electric field) that recruits and appears to orient the polysaccharide chains at the growing gel front. These studies demonstrate that short sequences (minutes) of low-power (∼1 V) electrical inputs can provide the program to guide self-assembly that yields hydrogels with stable, complex, and spatially varying structure and properties.
Morrow BH, Payne GF, and Shen J*
2015Stimuli-responsive self-assembly of polysaccharide through a rugged energy landscape.
J Am Chem Soc 137: 13024-13030, 2015
Surfactants
Morrow BH, Eike DM, Murch BP, Koenig PH, and Shen J*
2014Predicting proton titration in cationic micelle and bilayer environments.
J Chem Phys 141: 084714, 2014
Knowledge of the protonation behavior of pH-sensitive molecules in micelles and bilayers has significant implications in consumer product development and biomedical applications. However, the calculation of pKa's in such environments proves challenging using traditional structure-based calculations. Here we apply all-atom constant pH molecular dynamics with explicit ions and titratable water to calculate the pKa of a fatty acid molecule in a micelle of dodecyl trimethylammonium chloride and liquid as well as gel-phase bilayers of diethyl ester dimethylammonium chloride. Interestingly, the pKa of the fatty acid in the gel bilayer is 5.4, 0.4 units lower than that in the analogous liquid bilayer or micelle, despite the fact that the protonated carboxylic group is significantly more desolvated in the gel bilayer. This work illustrates the capability of all-atom constant pH molecular dynamics in capturing the delicate balance in the free energies of desolvation and Coulombic interactions. It also shows the importance of the explicit treatment of ions in sampling the protonation states. The ability to model dynamics of pH-responsive substrates in a bilayer environment is useful for improving fabric care products as well as our understanding of the side effects of anti-inflammatory drugs.
Morrow BH, Koenig PH, and Shen JK*
2013Self-Assembly and bilayer-micelle transition of fatty acids studied by replica-exchange constant pH molecular dynamics.
Langmuir 29: 14823-14830, 2013
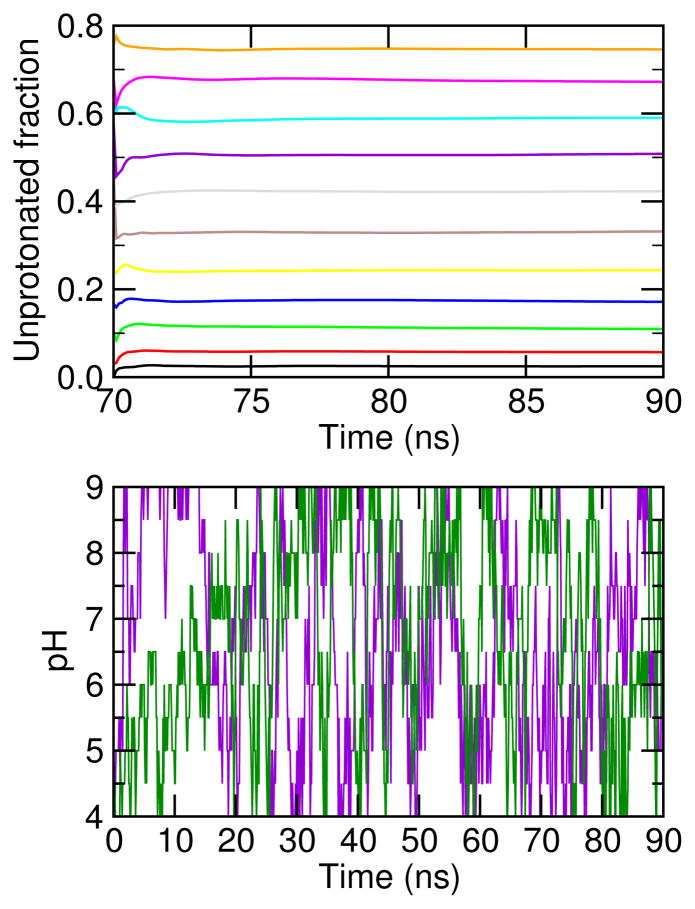
Recent interest in the development of surfactant-based nanodelivery systems targeting tumor sites has sparked our curiosity in understanding the detailed mechanism of the self-assembly and phase transitions of pH-sensitive surfactants. Toward this goal, we applied a state-of-the-art simulation technique, continuous constant pH molecular dynamics (CpHMD) with the hybrid-solvent scheme and pH-based replica-exchange protocol, to study the de novo self-assembly of 30 and 40 lauric acids, a simple model titratable surfactant. We observed the formation of a gel-state bilayer at low and intermediate pH and a spherical micelle at high pH, with the phase transition starting at 20-30% ionization and being completed at 50%. The degree of cooperativity for the transition increases from the 30-mer to the 40-mer. The calculated apparent or bulk pKa value is 7.0 for the 30-mer and 7.5 for the 40-mer. Congruent with experiment, these data demonstrate that CpHMD is capable of accurately modeling large conformational transitions of surfactant systems while allowing the simultaneous proton titration of constituent molecules. We suggest that CpHMD simulations may become a useful tool in aiding in the design and development of pH-sensitive nanocarriers for a variety of biomedical and technological applications.
Morrow BH, Koenig PH, and Shen JK*
2012Atomistic simulations of pH-dependent self-assembly of micelle and bilayer from fatty acids.
J Chem Phys 137: 194902, 2012

Detailed knowledge of the self-assembly and phase behavior of pH-sensitive surfactants has implications in areas such as targeted drug delivery. Here we present a study of the formation of micelle and bilayer from lauric acids using a state-of-the-art simulation technique, continuous constant pH molecular dynamics (CpHMD) with conformational sampling in explicit solvent and the pH-based replica-exchange protocol. We find that at high pH conditions a spherical micelle is formed, while at low pH conditions a bilayer is formed with a considerable degree of interdigitation. The mid-point of the phase transition is in good agreement with experiment. Preliminary investigation also reveals that the effect of counterions and salt screening shifts the transition mid-point and does not change the structure of the surfactant assembly. Based on these data we suggest that CpHMD simulations may be applied to computational design of surfactant-based nano devices in the future.
Morrow BH, Wang Y, Wallace JA, Koenig PH, and Shen JK*
2011Simulating pH titration of a single surfactant in ionic and nonionic micelles.
J Phys Chem B 115: 14980-14990, 2011
Calculation of surfactant pK(a)'s in micelles is a challenging task using traditional electrostatic methods due to the lack of structural data and information regarding the effective dielectric constant. Here we test the implicit- and hybrid-solvent-based continuous constant pH molecular dynamics (CpHMD) methods for predicting the pK(a) shift of a lauric acid solubilized in three micelles: dodecyl sulfate (DS), dodecyltrimethylammonium (DTA), and dodecyltriethylene glycol ether (DE3). Both types of simulations are able to reproduce the observed positive pK(a) shifts for the anionic DS and nonionic DE3 micelles. However, for the cationic DTA micelle, the implicit-solvent simulation fails to predict the direction of the pK(a) shift, while the hybrid-solvent simulation, where conformational sampling is conducted in explicit solvent, is consistent with experiment, although the specific-ion effects remain to be accurately determined. Comparison between the implicit- and hybrid-solvent data shows that the latter gives a more realistic description of the conformational environment of the titrating probe. Surprisingly, in the DTA micelle, surfactants are only slightly attracted to the laurate ion, which diminishes the magnitude of the electrostatic stabilization, resulting in a positive pK(a) shift that cannot be explained by chemical intuition or other theoretical models. Our data underscores the importance of microscopic models and ionization-coupled conformational dynamics in quantitative prediction of the pK(a) shifts in micelles.
Wang Y, Wallace JA, Koenig PH, and Shen JK*
2011Molecular dynamics simulations of ionic and nonionic surfactant micelles with a generalized Born implicit-solvent model.
J Comput Chem 32: 2358, 2011
In recent years, all-atom and coarse-grained models have been developed and applied to simulations of micelles and biological membranes. Here, we explore the question of whether a combined all-atom representation of surfactant molecules and continuum description of solvent based on the generalized Born model can be used to study surfactant micelles. Specifically, we report the parameterization of the GBSW model with a surface-area dependent nonpolar solvation energy term for dodecyl sulfate, dodecyl tetramethylammonium, and dodecyl triethyleneglycol ether molecules. In the parameterization procedure,the atomic Born radii were derived from the radial distribution functions of solvent charge and refined targeting the potential of mean force of dimer interactions from explicit-solvent simulations. The optimized radii were then applied in molecular dynamics simulations of the ionic and nonionic micelles.We found that the micelles are stable but more compact and rigid than in explicit solvent as a consequence of the drastic reduction in solvation and mobility of surfactant monomers within the micelle. Based on these data and our previous work, we suggest that in addition to a more accurate description of the nonpolar solvation energy, the ruggedness in the short-range interactions due to solvent granularity is a critical feature that needs to be taken into account to accurately model processes such as micelle formation and protein folding in implicit solvent. Finally, the explicit-solvent data presented here offers new insights into different conformational behavior of ionic and nonionic micelles which is valuable for understanding hydrophobic assemblies and of interest to the detergent industry.