Enzyme Dynamics and Inhibition
Ruibin Liu, Joseph Clayton, Mingzhe Shen, and Shen J*
2023Machine Learning Models for Interrogating Proteome-wide Cysteine Ligandabilities
Under review, 2023
PMCID: DOI: bioRxiv
Liu R, Vazquez-Montelongo EA, Ma S*, and Shen J*
2023Quantum Descriptors for Predicting and Understanding the Structure-Activity Relationships of Covalent Warheads
J Chem Inf Model 63: 4912-4923, 2023
PMCID: DOI: 10.1021/acs.jcim.3c00720
Romany E, Liu R, Zhan S, Clayton J, and Shen J*
2023Analysis of the ERK Pathway Cysteinome for Targeted Covalent Inhibition of RAF and MEK Kinases
J Chem Inf Model 63: 2483–2494, 2023
PMCID: PMCID DOI: 10.1021/acs.jcim.3c00014
Clayton J, de Oliveira V M, Ibrahim M F, Sun XY, Mahinthichaichan P, Shen M, Hilgenfeld R, Shen J*.
2023Integrative Approach to Dissect the Drug Resistance Mechanism of the H172Y Mutation of SARS-CoV-2 Main Protease
J Chem Inf Mode 63: 3521–3533, 2023
PMCID: PMC9387124 DOI: 10.1021/acs.jcim.3c00344
Yang R, Han S, Clayton C, Haghighatian M, Tsai C-C, Yao Y, Li P, Shen J, Zhou Q*
2022A proof-of-concept inhibitor of endothelial lipase suppresses triple-negative breast cancer cells by hijacking the mitochondrial function
Cancers 14: 3763, 2022
PMCID: PMC9367514 DOI: 10.3390/cancers14153763
Ma S*, Henderson JA, and Shen J*
2021Exploring the pH-dependent structure-dynamics-function relationship of human renin.
J Chem Inf Model 61: 400-407, 2021
PMCID: PMC7855609 DOI: 10.1021/acs.jcim.0c01201
Henderson JA and Shen J*
2022Exploring the pH- and Ligand-Dependent Flap Dynamics of Malarial Plasmepsin II.
J Chem Inf Model 62: 150–158, 2022
PMCID: PMC8812262 DOI: 10.1021/acs.jcim.1c01180
Verma N, Henderson JA, and Shen J*
2020Proton-Coupled Conformational Activation of SARS Coronavirus Main Proteases and Opportunity for Designing Small-Molecule Broad-Spectrum Targeted Covalent Inhibitors.
J Am Chem Soc 142: 21883–21890, 2020
Henderson JA, Verma N, and Shen J*
2020Assessment of proton-coupled conformational dynamics of SARS and MERS coronavirus papain-like proteases: Implication for designing broad-spectrum antiviral inhibitors
J Chem Phys, Special Issue “Classical Molecular Dynamics (MD) Simulations: Codes, Algorithms, Force Fields, and Applications”. In Press. Editor's Choice. AIP Press release, 2020
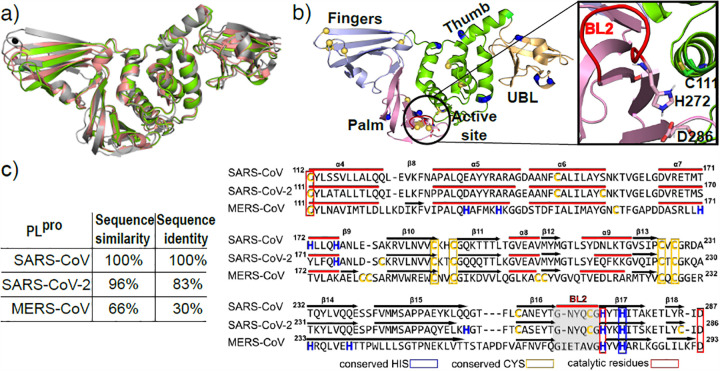
Broad-spectrum antiviral drugs are urgently needed to stop the COVID-19 pandemic and prevent future ones. The novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is related to SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV), which have caused the previous outbreaks. The papain-like protease (PLpro) is an attractive drug target due to its essential roles in the viral life cycle. As a cysteine protease, PLpro is rich in cysteines and histidines and their protonation/deprotonation modulates catalysis and conformational plasticity. Here we report the pK a calculations and assessment of the proton-coupled conformational dynamics of SARS-CoV-2 in comparison to SARS-CoV and MERS-CoV PLpros using a newly developed GPU-accelerated implicit-solvent continuous constant pH molecular dynamics method with an asynchronous replica-exchange scheme.
Harris RC, Liu R, and Shen J*
2020Predicting reactive cysteines with implicit-solvent based continuous constant pH molecular dynamics in Amber.
J Chem Theory Comput 16: 3689-3698, 2020
Cysteines existing in the deprotonated thiolate form or having a tendency to become deprotonated are important players in enzymatic and cellular redox functions and frequently exploited in covalent drug design; however, most computational studies assume cysteines as protonated. Thus, developing an efficient tool that can make accurate and reliable predictions of cysteine protonation states is timely needed. We recently implemented a generalized Born (GB) based continuous constant pH molecular dynamics (CpHMD) method in Amber for protein pKa calculations on CPUs and GPUs. Here we benchmark the performance of GB-CpHMD for predictions of cysteine pKa's and reactivities using a data set of 24 proteins with both down- and upshifted cysteine pKa's. We found that 10 ns single-pH or 4 ns replica-exchange CpHMD titrations gave root-mean-square errors of 1.2-1.3 and correlation coefficients of 0.8-0.9 with respect to experiment.
Tsai CC, Yue Z, and Shen J*
2019How electrostatic coupling enables conformational plasticity in a tyrosine kinase.
J Am Chem Soc 141: 15092-15101, 2019
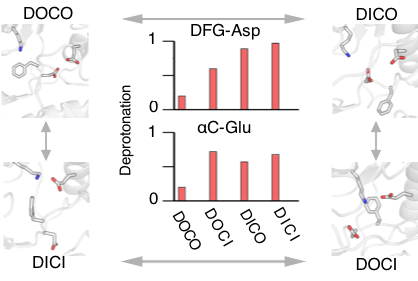
The ability to predictively model various conformational states could accelerate selectivity inhibitor design. We present, to our best knowledge, the first predictive computational study that captured the four major types of kinase conformations starting from a single crystal structure without employing a target structure, mutation, or bias. Our data revealed, for the first time, how protonation state changes of key residues facilitate the conformational transitions, providing a direct support and explanation for a long-standing hypothesis regarding the protonation-dependent DFG flip. Our simulations also uncovered intermediate states, among which a unique inactive state, which can be tested in selective inhibitor design.
Liu R, Yue Z, Tsai CC, and Shen J*
2019Assessing lysine and cysteine reactivities for designing targeted covalent kinase inhibitors.
J Am Chem Soc 141: 6553-6560, 2019
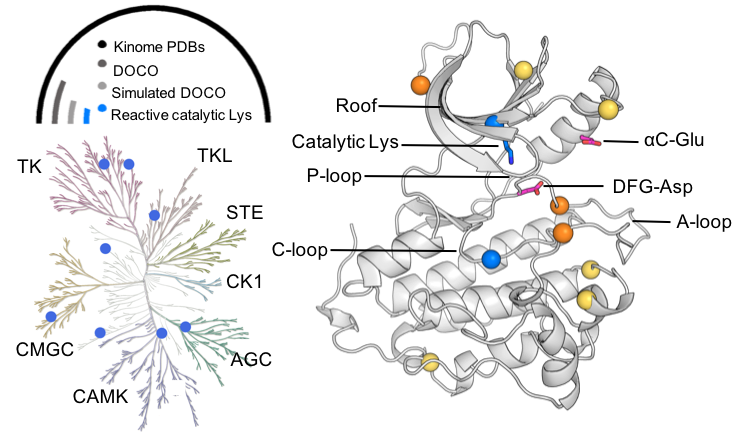
We present a computational tool to assess the nucleophilicities of lysines and cysteines to accelerate covalent drug design. The tool enabled us to discover that kinases in a rare inactive conformation can be covalently targeted at catalytic lysines. A new strategy for designing highly selective, lysine-targeted covalent kinase inhibitors was proposed to address the emergent drug resistance issue facing the cysteine-targeted strategy and help usher in the next generation covalent kinase inhibitor design.
Henderson JA, Harris RC, Tsai CC, and Shen J*
2018How ligand protonation proton state controls water in protein-ligand binding.
J Phys Chem Lett 9: 5440-5444, 2018
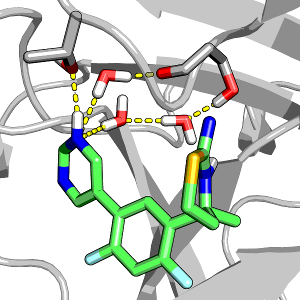
The role of water in protein-ligand has been intensely studied in recent years; however, how ligand protonation state change perturbs water has not been investigated. Here we use two homologues aspartyl proteases to illustrate how modeling protein-ligand binding while allowing ligand titration can further advance our understanding of the role of water in modulating the inhibitor selectivity for a particular target.
Huang YD, Yue Z, Tsai CC, Henderson JA, and Shen J*
2018Predicting catalytic proton donors and nucleophiles in enzymes: how adding dynamics helps elucidate the structure-function relationships.
J Phys Chem Lett 9: 1179-1184, 2018
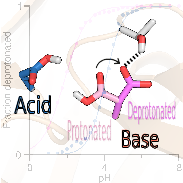
Despite the importance, prediction of catalytic proton donors and nucleophiles has remained an unsolved problem. Here we tested three types of state-of-the-art computational methods to predict the pKa’s of buried and coupled carboxyl dyads in enzymes. We asked the question what makes a residue proton donor vs. nucleophile. Our study revealed a surprisingly simple trend, which is not apparent from crystal structures, empirical or traditional pKa calculations.
Harris RC, Tsai CC, Ellis CR, Shen J*
2017Proton-coupled conformational allostery modulates the inhibitor selectivity for β-secretase.
J Phys Chem Lett 8: 4832-4837, 2017
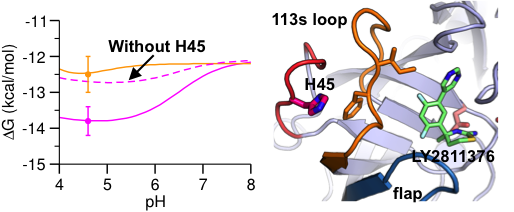
Many pharmaceutical targets, such as aspartyl proteases and kinases, exhibit pH-dependent dynamics and functions. Accurate prediction of their binding free energies is challenging, and current computational studies neglect the effects of pH and changes in protonation states. We demonstrated a new protocol to improve the predictive power of current binding free energy calculations. The allosteric mechanism discovered here could lead to exciting opportunities in drug design.
Ellis CR, Tsai CC, Lin FY, and Shen J*
2017Conformational dynamics of cathepsin D and binding to a small-molecule BACE1 inhibitor.
J Comput Chem 38: 1260-1269, 2017
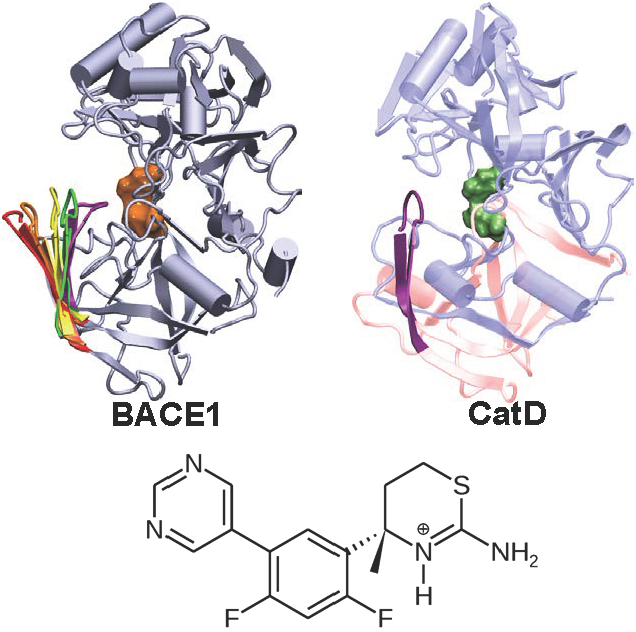
Targeting the aspartyl protease BACE1 with small-molecule inhibitors offers a promising route for treatment of Alzheimer's disease. However, developing inhibitors that can selectively target BACE1 in favor of other proteases, especially cathepsin D, has presented significant challenges. We reported the first computational study that characterizes the conformational dynamics of capthesin D and how it binds a BACE1 small-molecule inhibitor.
Ellis CR, Tsai CC, Hou XJ, and Shen J*
2016Constant pH molecular dynamics reveals pH-modulated binding of two small-molecule BACE1 inhibitors.
J Phys Chem Lett 7: 944-949, 2016
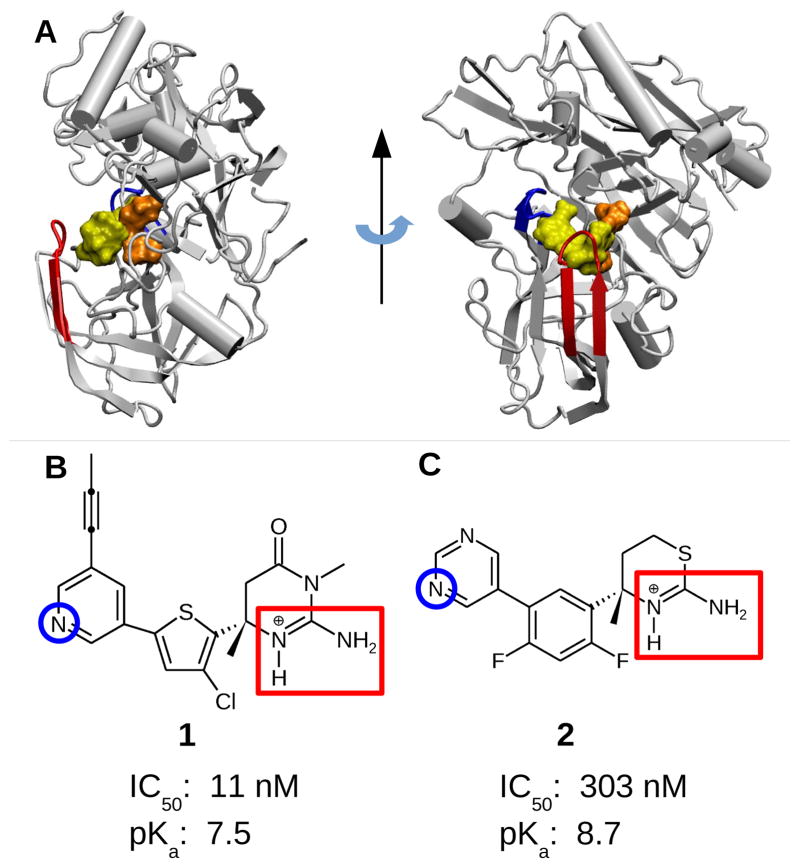
Targeting β-secretase (BACE1) with small-molecule inhibitors offers a promising route for treatment of Alzheimer's disease. However, the intricate pH dependence of BACE1 function and inhibitor efficacy has posed major challenges for structure-based drug design. Here we investigate two structurally similar BACE1 inhibitors that have dramatically different inhibitory activity using continuous constant pH molecular dynamics (CpHMD). At high pH, both inhibitors are stably bound to BACE1; however, within the enzyme active pH range, only the iminopyrimidinone-based inhibitor remains bound, while the aminothiazine-based inhibitor becomes partially dissociated following the loss of hydrogen bonding with the active site and change of the 10s loop conformation.
Ellis CR and Shen J*
2015pH-dependent population shift regulates BACE1 activity and inhibition.
J Am Chem Soc 137: 9543-9546, 2015
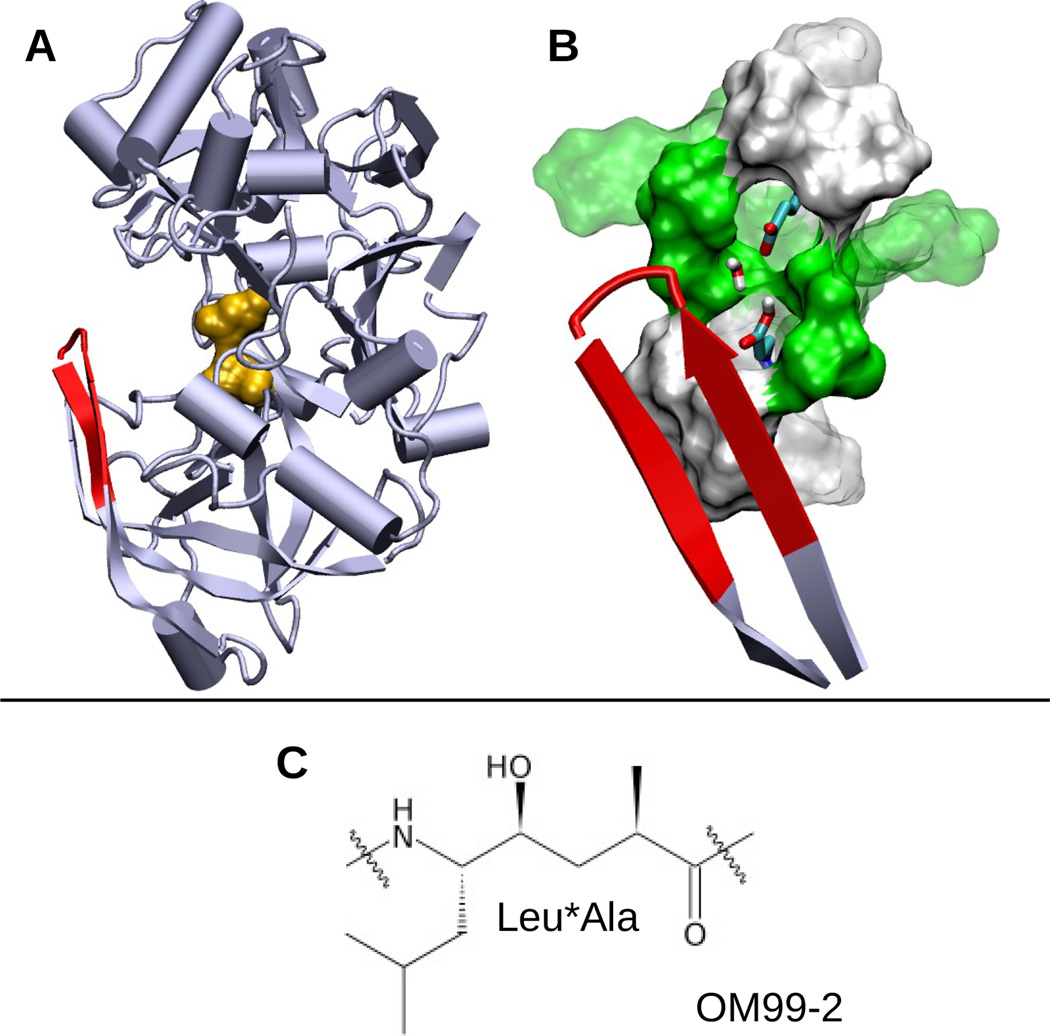
BACE1, a major therapeutic target for treatment of Alzheimer's disease, functions within a narrow pH range. Despite tremendous effort and progress in the development of BACE1 inhibitors, details of the underlying pH-dependent regulatory mechanism remain unclear. Here we elucidate the pH-dependent conformational mechanism that regulates BACE1 activity using continuous constant-pH molecular dynamics (MD). The simulations reveal that BACE1 mainly occupies three conformational states and that the relative populations of the states shift according to pH.