Our materials projects were motivated by questions from our partners in industry and engineering. For example, knowledge of the pKa’s of surfactants in various environments would be valuable for detergent formulation; however, the lack of structure makes the problem extremely challenging if possible at all for traditional structure-based continuum methods. We published the first study showing that the pKa's of surfactant micelles can be accurately calculated using hybrid-solvent CpHMD. Later, we also published the first study that calculates surfactant phase transition pKa’s. More recently, we extended the method to study self-assembly and dynamics of polysaccharide-based hydrogels using all-atom CpHMD.
Polysaccharide-based hydrogel films
Romany A, Payne GF, Shen J*
2023Mechanism of the Temperature-Dependent Self-Assembly and Polymorphism of Chitin
Chem Mater 35: 6472–6481, 2023
PMCID: DOI: 10.1021/acs.chemmater.3c01313
Liu Y, Kim E, Lei M, Wu S, Yan K, Shen J, Bentley W, Qu X, Shi X, Payne, GF*
2023Electro-bio-fabrication: Coupling Electrochemical and Biomolecular Methods to Create Functional Bio-based Hydrogels
Biomacromolecules 24: 2409-2432, 2023
PMCID: DOI: 10.1021/acs.biomac.3c00132
Mahinthichaichan P, Tsai CC, Payne, GF, and Shen J*
2020Polyelectrolytes in electric field: disparate conformational behavior along an amino-polysaccharide chain.
ACS Omega 5: 12016-12026, 2020
PMCID: PMC7271050 DOI: 10.1021/acsomega.0c00164
Wu S, Shi X, Yan K, Li J, Huynh R, Raub C, Shen J and Payne GF*
2020Electrical cuing of chitosan's mesoscale organization.
React Funct Polym 148: 104492, 2020
PMCID: None DOI: 10.1016/j.reactfunctpolym.2020.104492
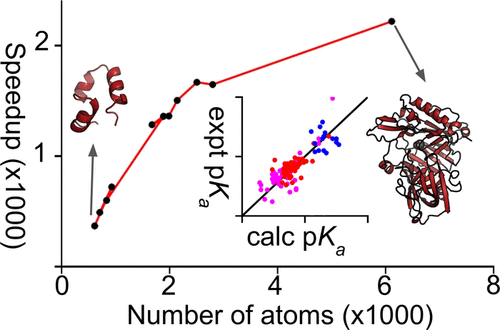
We present a GPU implementation of the continuous constant pH molecular dynamics (CpHMD) based on the most recent generalized Born implicit-solvent model in the pmemd engine of the Amber molecular dynamics package. To test the accuracy of the tool for rapid pKa predictions, a series of 2 ns single-pH simulations were performed for over 120 titratable residues in 10 benchmark proteins that were previously used to test the various continuous CpHMD methods.
Tsai CC, Payne GF, and Shen J*
2018Exploring pH-responsive, switchable crosslinking mechanisms for programming reconfigurable hydrogels based on aminopolysaccharides.
Chem Mater 30, 8597-860, 2018
Dynamic, stimuli-responsive, reconfigurable materials are finding growing interest and applications. A recent experiment (He et al., Adv Funct Matter, 2017) demonstrated a pH-responsive, reconfigurable hydrogel film based on a single biopolymer chitosan and two physical crosslinking mechanisms. Here we use state-of-the-art molecular dynamics simulations to gain atomically-detailed insights into the three salient features of this experimental system: 1) a pH-responsive switch between the crystalline network junctions formed by intermolecular hydrogen bonding between chitosan chains and electrostatic crosslinks based on sodium dodecyl sulfate (SDS) micelles; 2) viscoelastic behavior of the SDS-crosslinked network; and 3) a stable but erasable gradient between the two crosslinked regions.
Wu S, Yan K, Zhao Y, Tsai CC, Shen J, Bentley WE, Chen Y, Deng H, Du Y, Payne GF* and Shi X*
2018Electrical writing onto a dynamically responsive polysaccharide medium: patterning structure and function into a reconfigurable medium.
Adv Funct Mater, 28: 1803139, 2018
Historically, paper is the medium to write information. Here, the concept of writing is expanded from the addition of mass (i.e., ink) onto a static polysaccharide medium (i.e., paper), to the addition of electrical energy to a dynamically reconfigurable polysaccharide medium. Specifically, a dual‐responsive interpenetrating polysaccharide network is used as a dynamic medium. Electrical writing is achieved using an electrode pen to locally perform the cathodic electrolysis reactions that generate high‐pH regions that neutralize the interpenetrating chitosan chains and induce their self‐assembly into crystalline regions. Surprisingly, the gradients in structure induced by cathodic writing are stable even after the pH gradient has dissipated.
Yan K, Liu Y, Zhang J, Correa SO, Shang W, Tsai CC, Bentley WE, Shen J, Scarcelli G, Raub CB, Shi XW, Payne GF*
2018Electrical programming of soft matter: using temporally varying electrical inputs to spatially control self assembly.
Biomacromolecules 19: 364-373, 2018
The growing importance of hydrogels in translational medicine has stimulated the development of top-down fabrication methods, yet often these methods lack the capabilities to generate the complex matrix architectures observed in biology. Here we show that temporally varying electrical signals can cue a self-assembling polysaccharide to controllably form a hydrogel with complex internal patterns. Evidence from theory and experiment indicate that internal structure emerges through a subtle interplay between the electrical current that triggers self-assembly and the electrical potential (or electric field) that recruits and appears to orient the polysaccharide chains at the growing gel front. These studies demonstrate that short sequences (minutes) of low-power (∼1 V) electrical inputs can provide the program to guide self-assembly that yields hydrogels with stable, complex, and spatially varying structure and properties.
Tsai CC, Morrow BH, Chen W, Payne GP, and Shen J*
Towards understanding the environmental control of hydrogel film properties: how salt modulates the flexibility of chitosan chains.
Macromolecules 50: 5946-5952, 2017
Chitosan is a pH-responsive self-assembling polysaccharide that can be electrodeposited to form a hydrogel film. During electrodeposition a host of dynamic processes occur simultaneously over a hierarchy of length scales. Experiments have shown that the microstructure and properties of the deposited gels are highly dependent on the solution conditions and imposed electrical signals. However, detailed mechanisms are not understood. Here we use molecular dynamics simulations to explore the conformational dynamics of individual chitosan chains composed of 20 glucosamine units in aqueous solution. Simulations yield a total persistence length of about 5 nm, in agreement with the lower range of the experimental estimates and supporting a worm-like-chain model. The surprisingly high flexibility arises from the glycosidic linkage sampling an appreciable population of anti-ψ conformation which is associated with backbone bending and higher flexibility. Significantly, the presence of added salt increases the anti-ψ population and consequently flexibility for both charged and neutral chains. The latter is due to counterion binding and disruption of an intramolecular hydrogen bond that stabilizes the extended conformation. Thus, our data suggest that salt concentration modulates the conformation and dynamics of individual chitosan chains in addition to interchain hydrogen bonding during pH-induced self-assembly. The insight from this work provides a missing piece of the puzzle toward understanding the complex mechanism by which solution conditions control the hydrogel properties of chitosan at the microscopic level which cannot be accessed experimentally.
Kim E, Liu Y, Ben-Yoav H, Winkler TE, Yan K, Shi S, Shen J, Kelly DL, Ghodssi R, Bentley WE, and Payne GF*
2016Fusing sensor paradigms to acquire chemical information: an integrative role for smart biopolymeric hydrogels.
Adv Healthcare Mater 5: 2595-2616, 2016
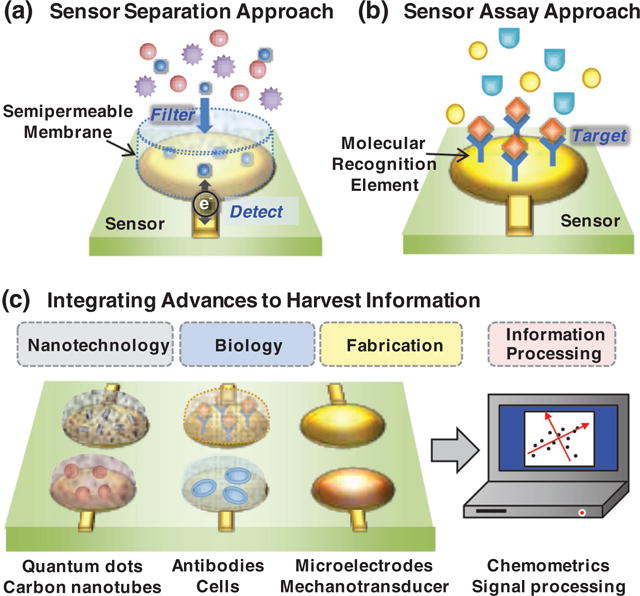
The Information Age transformed our lives but it has had surprisingly little impact on the way chemical information (e.g., from our biological world) is acquired, analyzed and communicated. Sensor systems are poised to change this situation by providing rapid access to chemical information. This access will be enabled by technological advances from various fields: biology enables the synthesis, design and discovery of molecular recognition elements as well as the generation of cell-based signal processors; physics and chemistry are providing nano-components that facilitate the transmission and transduction of signals rich with chemical information; microfabrication is yielding sensors capable of receiving these signals through various modalities; and signal processing analysis enhances the extraction of chemical information. The authors contend that integral to the development of functional sensor systems will be materials that (i) enable the integrative and hierarchical assembly of various sensing components (for chemical recognition and signal transduction) and (ii) facilitate meaningful communication across modalities. It is suggested that stimuli-responsive self-assembling biopolymers can perform such integrative functions, and redox provides modality-spanning communication capabilities. Recent progress toward the development of electrochemical sensors to manage schizophrenia is used to illustrate the opportunities and challenges for enlisting sensors for chemical information processing.
Kim E, Xing Y, Cheng Y, Wu H-C, Liu Y, Morrow BH, Ben-Yoav H, Ghodssi R, Rubloff GW, Shen J, Bentley WE, Shi X, and Payne GF*
2016Stimuli-responsive self-assembly of polysaccharide through a rugged energy landscape.
J Am Chem Soc 137: 13024-13030, 2015
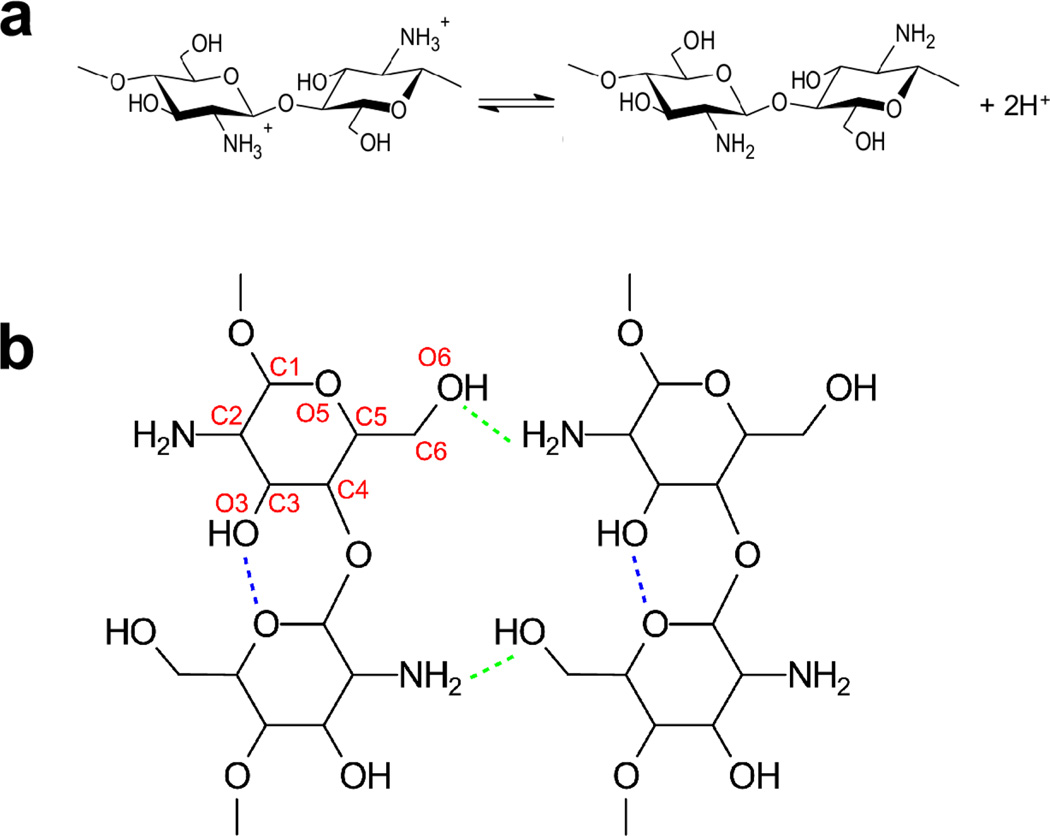
Self-assembling polysaccharides can form complex networks with structures and properties highly dependent on the sequence of triggering cues. Controlling the emergence of such networks provides an opportunity to create soft matter with unique features; however, it requires a detailed understanding of the subtle balance between the attractive and repulsive forces that drives the stimuli-induced self-assembly. Here we employ all-atom molecular dynamics simulations on the order of 100 ns to study the mechanisms of the pH-responsive gelation of the weakly basic aminopolysaccharide chitosan. We find that low pH induces a sharp transition from gel to soluble state, analogous to pH-dependent folding of proteins, while at neutral and high pH self-assembly occurs via a rugged energy landscape, reminiscent of RNA folding. A surprising role of salt is to lubricate the conformational search for the thermodynamically stable states. Although our simulations represent the early events in the self-assembly process of chitosan, which may take seconds or minutes to complete, the atomically detailed insights are consistent with recent experimental observations and provide a basis for understanding how environmental conditions modulate the structure and mechanical properties of the self-assembled polysaccharide systems. The ability to control structure and properties via modification of process conditions will aid in the technological efforts to create complex soft matter with applications ranging from bioelectronics to regenerative medicine.
Kim E, Xing Y, Cheng Y, Wu H-C, Liu Y, Morrow BH, Ben-Yoav H, Ghodssi R, Rubloff GW, Shen J, Bentley WE, Shi X, and Payne GF*
2016Chitosan to connect biology to electronics: Fabricating the bio-device interface and communicating across this interface.
Polymers 7: 1-46, 2015
Self-assembling polysaccharides can form complex networks with structures and properties highly dependent on the sequence of triggering cues. Controlling the emergence of such networks provides an opportunity to create soft matter with unique features; however, it requires a detailed understanding of the subtle balance between the attractive and repulsive forces that drives the stimuli-induced self-assembly. Here we employ all-atom molecular dynamics simulations on the order of 100 ns to study the mechanisms of the pH-responsive gelation of the weakly basic aminopolysaccharide chitosan. We find that low pH induces a sharp transition from gel to soluble state, analogous to pH-dependent folding of proteins, while at neutral and high pH self-assembly occurs via a rugged energy landscape, reminiscent of RNA folding. A surprising role of salt is to lubricate the conformational search for the thermodynamically stable states. Although our simulations represent the early events in the self-assembly process of chitosan, which may take seconds or minutes to complete, the atomically detailed insights are consistent with recent experimental observations and provide a basis for understanding how environmental conditions modulate the structure and mechanical properties of the self-assembled polysaccharide systems. The ability to control structure and properties via modification of process conditions will aid in the technological efforts to create complex soft matter with applications ranging from bioelectronics to regenerative medicine.
Peptide-based Nanofiber
Cote Y, Fu IW, Dobson ET, Goldberger JE, Nguyen HD, and Shen J*
2014Mechanism of the pH-controlled self-assembly of nanofibers from peptide amphiphiles.
J Phys Chem C 118: 16272-16278, 2014

Stimuli-responsive, self-assembling nanomaterials hold a great promise to revolutionize medicine and technology. However, current discovery is slow and often serendipitous. Here we report a multiscale modeling study to elucidate the pH-controlled self-assembly of nanofibers from the peptide amphiphiles, palmitoyl-I-A3E4-NH2. The coarse-grained simulations revealed the formation of random-coil based spherical micelles at strong electrostatic repulsion. However, at weak or no electrostatic repulsion, the micelles merge into a nanofiber driven by the β-sheet formation between the peptide segments. The all-atom constant pH molecular dynamics revealed a cooperative transition between random coil and β-sheet in the pH range 6-7, matching the CD data. Interestingly, although the bulk pKa is more than one unit below the transition pH, consistent with the titration data, the highest pKa's coincide with the transition pH, suggesting that the latter may be tuned by modulating the pKa's of a few solvent-buried Glu side chains. Together, these data offer, to our best knowledge, the first multiresolution and quantitative view of the pH-dependent self-assembly of nanofibers. The novel protocols and insights gained are expected to advance the computer-aided design and discovery of pH-responsive nanomaterials.
Surfactants for detergents
Morrow BH, Eike DM, Murch BP, Koenig PH, and Shen J*
2014Predicting proton titration in cationic micelle and bilayer environments.
J Chem Phys 141: 084714, 2014
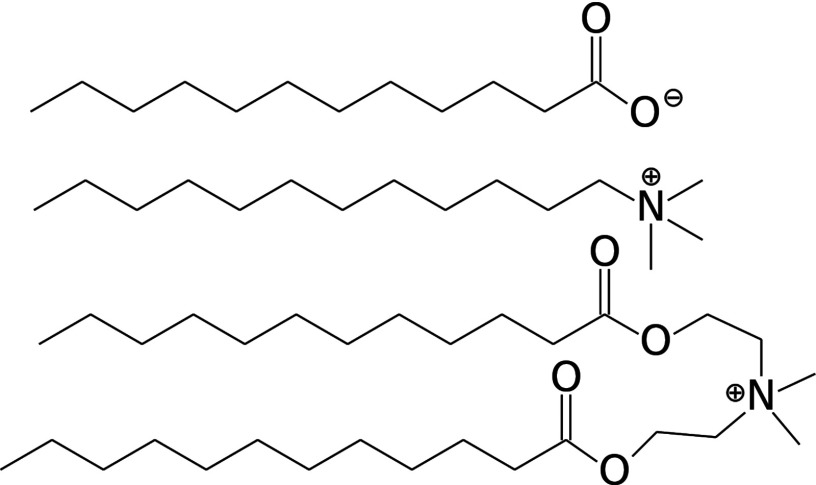
Knowledge of the protonation behavior of pH-sensitive molecules in micelles and bilayers has significant implications in consumer product development and biomedical applications. However, the calculation of pKa's in such environments proves challenging using traditional structure-based calculations. Here we apply all-atom constant pH molecular dynamics with explicit ions and titratable water to calculate the pKa of a fatty acid molecule in a micelle of dodecyl trimethylammonium chloride and liquid as well as gel-phase bilayers of diethyl ester dimethylammonium chloride. Interestingly, the pKa of the fatty acid in the gel bilayer is 5.4, 0.4 units lower than that in the analogous liquid bilayer or micelle, despite the fact that the protonated carboxylic group is significantly more desolvated in the gel bilayer. This work illustrates the capability of all-atom constant pH molecular dynamics in capturing the delicate balance in the free energies of desolvation and Coulombic interactions. It also shows the importance of the explicit treatment of ions in sampling the protonation states. The ability to model dynamics of pH-responsive substrates in a bilayer environment is useful for improving fabric care products as well as our understanding of the side effects of anti-inflammatory drugs.
Morrow BH, Koenig PH, and Shen JK*
2013Self-Assembly and bilayer-micelle transition of fatty acids studied by replica-exchange constant pH molecular dynamics.
Langmuir 29: 14823-14830, 2013
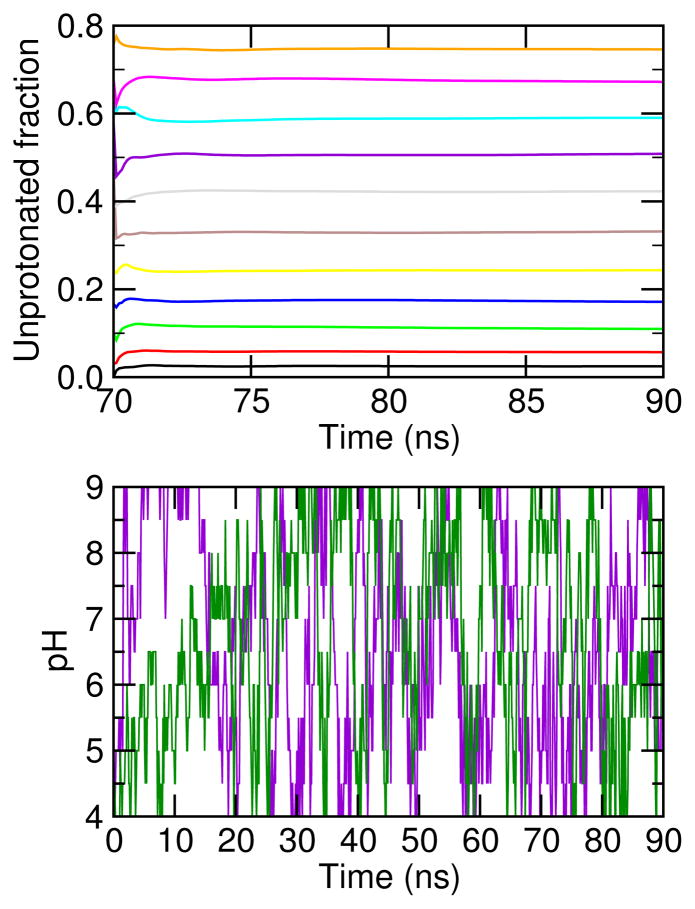
Recent interest in the development of surfactant-based nanodelivery systems targeting tumor sites has sparked our curiosity in understanding the detailed mechanism of the self-assembly and phase transitions of pH-sensitive surfactants. Toward this goal, we applied a state-of-the-art simulation technique, continuous constant pH molecular dynamics (CpHMD) with the hybrid-solvent scheme and pH-based replica-exchange protocol, to study the de novo self-assembly of 30 and 40 lauric acids, a simple model titratable surfactant. We observed the formation of a gel-state bilayer at low and intermediate pH and a spherical micelle at high pH, with the phase transition starting at 20-30% ionization and being completed at 50%. The degree of cooperativity for the transition increases from the 30-mer to the 40-mer. The calculated apparent or bulk pKa value is 7.0 for the 30-mer and 7.5 for the 40-mer. Congruent with experiment, these data demonstrate that CpHMD is capable of accurately modeling large conformational transitions of surfactant systems while allowing the simultaneous proton titration of constituent molecules. We suggest that CpHMD simulations may become a useful tool in aiding in the design and development of pH-sensitive nanocarriers for a variety of biomedical and technological applications.
Morrow BH, Koenig PH, and Shen JK*
2012Atomistic simulations of pH-dependent self-assembly of micelle and bilayer from fatty acids.
J Chem Phys 137: 194902, 2012

Detailed knowledge of the self-assembly and phase behavior of pH-sensitive surfactants has implications in areas such as targeted drug delivery. Here we present a study of the formation of micelle and bilayer from lauric acids using a state-of-the-art simulation technique, continuous constant pH molecular dynamics (CpHMD) with conformational sampling in explicit solvent and the pH-based replica-exchange protocol. We find that at high pH conditions a spherical micelle is formed, while at low pH conditions a bilayer is formed with a considerable degree of interdigitation. The mid-point of the phase transition is in good agreement with experiment. Preliminary investigation also reveals that the effect of counterions and salt screening shifts the transition mid-point and does not change the structure of the surfactant assembly. Based on these data we suggest that CpHMD simulations may be applied to computational design of surfactant-based nano devices in the future.
Morrow BH, Wang Y, Wallace JA, Koenig PH, and Shen JK*
2011Simulating pH titration of a single surfactant in ionic and nonionic micelles.
J Phys Chem B 115: 14980-14990, 2011
Calculation of surfactant pK(a)'s in micelles is a challenging task using traditional electrostatic methods due to the lack of structural data and information regarding the effective dielectric constant. Here we test the implicit- and hybrid-solvent-based continuous constant pH molecular dynamics (CpHMD) methods for predicting the pK(a) shift of a lauric acid solubilized in three micelles: dodecyl sulfate (DS), dodecyltrimethylammonium (DTA), and dodecyltriethylene glycol ether (DE3). Both types of simulations are able to reproduce the observed positive pK(a) shifts for the anionic DS and nonionic DE3 micelles. However, for the cationic DTA micelle, the implicit-solvent simulation fails to predict the direction of the pK(a) shift, while the hybrid-solvent simulation, where conformational sampling is conducted in explicit solvent, is consistent with experiment, although the specific-ion effects remain to be accurately determined. Comparison between the implicit- and hybrid-solvent data shows that the latter gives a more realistic description of the conformational environment of the titrating probe. Surprisingly, in the DTA micelle, surfactants are only slightly attracted to the laurate ion, which diminishes the magnitude of the electrostatic stabilization, resulting in a positive pK(a) shift that cannot be explained by chemical intuition or other theoretical models. Our data underscores the importance of microscopic models and ionization-coupled conformational dynamics in quantitative prediction of the pK(a) shifts in micelles.
Wang Y, Wallace JA, Koenig PH, and Shen JK*
2011Molecular dynamics simulations of ionic and nonionic surfactant micelles with a generalized Born implicit-solvent model.
J Comput Chem 32: 2358, 2011
In recent years, all-atom and coarse-grained models have been developed and applied to simulations of micelles and biological membranes. Here, we explore the question of whether a combined all-atom representation of surfactant molecules and continuum description of solvent based on the generalized Born model can be used to study surfactant micelles. Specifically, we report the parameterization of the GBSW model with a surface-area dependent nonpolar solvation energy term for dodecyl sulfate, dodecyl tetramethylammonium, and dodecyl triethyleneglycol ether molecules. In the parameterization procedure,the atomic Born radii were derived from the radial distribution functions of solvent charge and refined targeting the potential of mean force of dimer interactions from explicit-solvent simulations. The optimized radii were then applied in molecular dynamics simulations of the ionic and nonionic micelles.We found that the micelles are stable but more compact and rigid than in explicit solvent as a consequence of the drastic reduction in solvation and mobility of surfactant monomers within the micelle. Based on these data and our previous work, we suggest that in addition to a more accurate description of the nonpolar solvation energy, the ruggedness in the short-range interactions due to solvent granularity is a critical feature that needs to be taken into account to accurately model processes such as micelle formation and protein folding in implicit solvent. Finally, the explicit-solvent data presented here offers new insights into different conformational behavior of ionic and nonionic micelles which is valuable for understanding hydrophobic assemblies and of interest to the detergent industry.