Development of Continuous Constant pH Molecular Dynamics
In recent years, Shen lab further developed CpHMD to improve the accuracy and generalizability. The developments includes the GB-Neck2 based CpHMD, the hybrid-solvent and all-atom GRF and PME CpHMD. Our goal is to offer an accurate, reliable and computational efficient pH-controlling tool (pH stat) to the community in the near future. The CpHMD tools are implemented in both CHARMM and Amber packages. We have recently completed the GPU implementation of the GBNeck2-CpHMD method in Amber. The GPU implementation of the all-atom PME-CpHMD method in Amber is under testing.
de Oliveira VM, Liu R, Shen J*
2022Constant pH Molecular Dynamics Simulations: Current Status and Recent Applications
Curr Opin Struct Biol 77: 102498, 2022
PMCID: PMC9933785 DOI: 10.1016/j.sbi.2022.102498
Harris, JA, Liu, R, Martins de Oliveira, V, Vazquez Montelongo, E, Henderson, JA, and Shen, J
2022GPU-Accelerated All-atom Particle-Mesh Ewald Continuous Constant pH Molecular Dynamics in Amber
J Chem Theory Comput 18: 7510–7527, 2022
PMCID: PMC10130738 DOI: 10.1021/acs.jctc.2c00586
Liu R, Henderson JA, Harris JA, Huang Y, de Oliveira MV, and Shen J*
2022A Guide to the Continuous Constant pH Molecular Dynamics Methods in Amber and CHARMM [v1.0]
Liv J Comput Mol Sci 4: 1563, 2022
PMCID: PMC9910290 DOI: 10.33011/livecoms.4.1.1563
Harris RC and Shen J*
2019GPU-accelerated implementation of continuous constant pH molecular dynamics in Amber: pKa predictions with single pH simulations.
J Chem Inf Model 59: 4821-4832, 2019
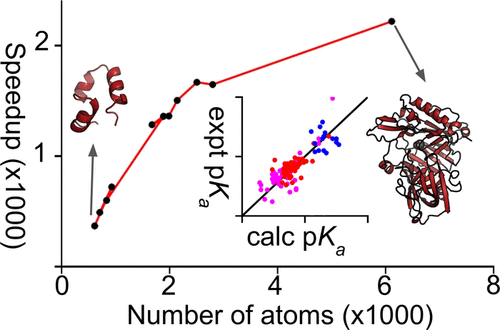
We present a GPU implementation of the continuous constant pH molecular dynamics (CpHMD) based on the most recent generalized Born implicit-solvent model in the pmemd engine of the Amber molecular dynamics package. To test the accuracy of the tool for rapid pKa predictions, a series of 2 ns single-pH simulations were performed for over 120 titratable residues in 10 benchmark proteins that were previously used to test the various continuous CpHMD methods.
Huang YD, Harris RC, Shen J*
2018Generalized Born based continuous constant pH molecular dynamics in Amber: implementation, benchmarking, and analysis.
J Chem Inf Model 58:1372-1383, 2018

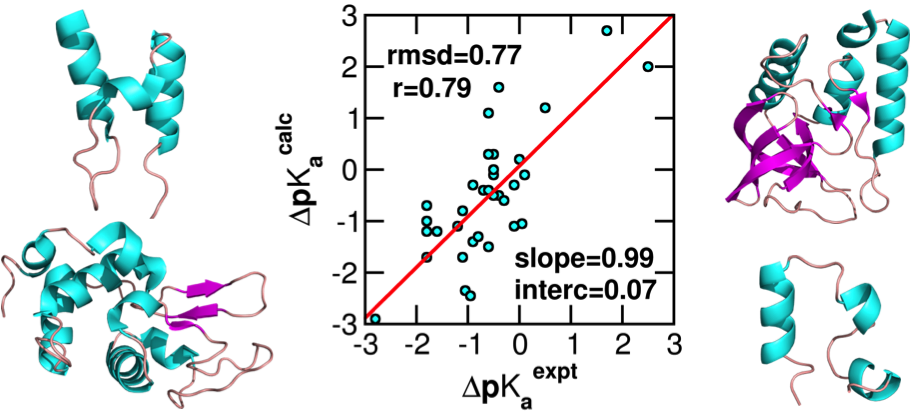
We report the CPU implementation of the CpHMD method based on the GBNeck2 generalized Born (GB) implicit-solvent model in the pmemd engine of the Amber molecular dynamics package. Benchmark data based on 11 proteins are discussed. The related software is disseminated through Amber18.
Huang YD, Chen W, and Shen J*
2016Continuous constant pH molecular dynamics with particle-mesh Ewald and titratabe water.
Chem Theory Comput 12: 5411-5421, 2016
Development of a pH stat to control solution pH in biomolecular simulations remains a outstanding goal. In recent years, the constant pH molecular dynamics methods have emerged; however, the accuracy and generality have been hampered by the use of implicit-solvent models or truncation-based electrostatic schemes. We reported the first implementation of the all-atom PME-based continuous constant pH method and benchmark results on a set of commonly used proteins.
Chen W and Shen J*
2014Effects of system net charge and electrostatic truncation on all-atom constant pH molecular dynamics.
J Comput Chem 35: 1986-1996, 2014
Constant pH molecular dynamics offers a means to rigorously study the effects of solution pH on dynamical processes. Here, we address two critical questions arising from the most recent developments of the all-atom continuous constant pH molecular dynamics (CpHMD) method: (1) What is the effect of spatial electrostatic truncation on the sampling of protonation states? (2) Is the enforcement of electrical neutrality necessary for constant pH simulations?
Chen W, Wallace JA, Yue Z, and Shen JK*
2013Introducing titratable water to all-atom molecular dynamics at constant pH.
Biophys J 105: L15-L17, 2013
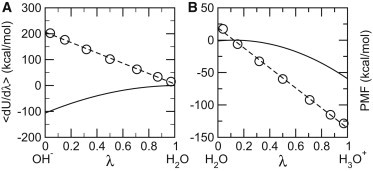
Recent development of titratable coions has paved the way for realizing all-atom molecular dynamics at constant pH. To further improve physical realism, here we describe a technique in which proton titration of the solute is directly coupled to the interconversion between water and hydroxide or hydronium. We test the new method in replica-exchange continuous constant pH molecular dynamics simulations of three proteins, HP36, BBL, and HEWL.
Wallace JA and Shen JK*
2012Charge-leveling and proper treatment of long-range electrostatics in all-atom molecular dynamics at constant pH.
J Chem Phys 137: 184105, 2012
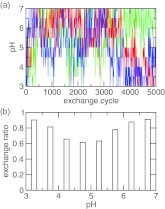
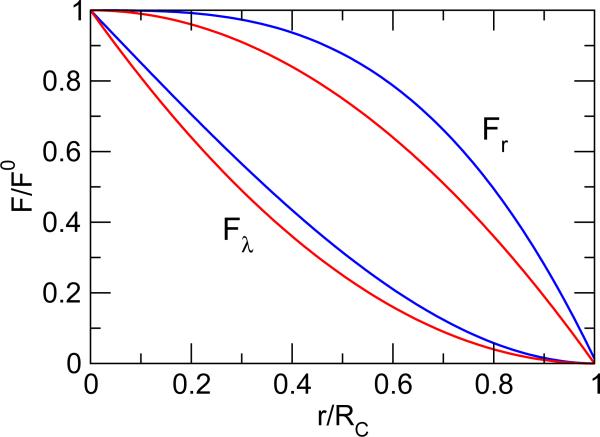
Recent development of constant pH molecular dynamics (CpHMD) methods has offered promise for adding pH-stat in molecular dynamics simulations. However, until now the working pH molecular dynamics (pHMD) implementations are dependent in part or whole on implicit-solvent models. Here we show that proper treatment of long-range electrostatics and maintaining charge neutrality of the system are critical for extending the continuous pHMD framework to the all-atom representation. The former is achieved here by adding forces to titration coordinates due to long-range electrostatics based on the generalized reaction field method, while the latter is made possible by a charge-leveling technique that couples proton titration with simultaneous ionization or neutralization of a co-ion in solution. We test the new method using the pH-replica-exchange CpHMD simulations of a series of aliphatic dicarboxylic acids with varying carbon chain length.
The first-generation CpHMD technique was developed in the Brooks group. This method utilizes the generalized Born implicit-solvent model for both conformational and protonation state sampling. It was recognized as one of the most accurate pKa prediction tools in the blind protein pKa prediction exercise in 2009.
Wallace JA and Shen JK*
2011Continuous constant pH molecular dynamics in explicit solvent with pH-based replica exchange.
J Chem Theory Comput 7: 2617-2629, 2011
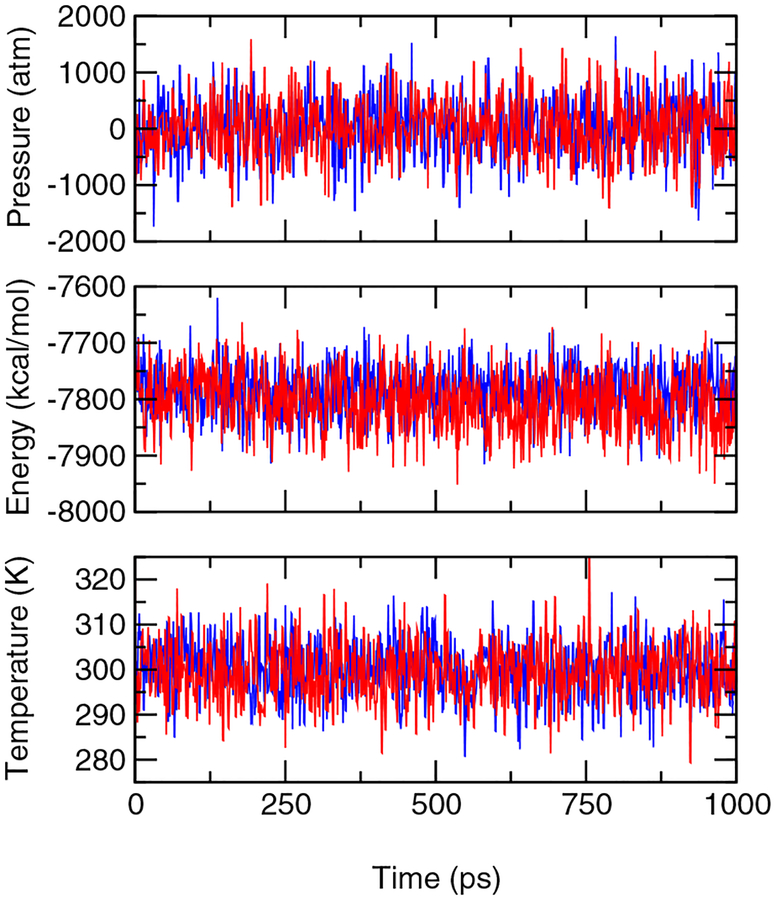
A computational tool that offers accurate pKa values and atomically detailed knowledge of protonation-coupled conformational dynamics is valuable for elucidating mechanisms of energy transduction processes in biology, such as enzyme catalysis and electron transfer as well as proton and drug transport. Toward this goal we present a new technique of embedding continuous constant pH molecular dynamics within an explicit-solvent representation. In this technique we make use of the efficiency of the generalized-Born (GB) implicit-solvent model for estimating the free energy of protein solvation while propagating conformational dynamics using the more accurate explicit-solvent model. Also, we employ a pH-based replica exchange scheme to significantly enhance both protonation and conformational state sampling.
Khandogin J and Brooks CL III*
2006Toward the accurate first-principles prediction of ionization equilibria in proteins.
Biochemistry 45: 9363-9373, 2006
The calculation of pK(a) values for ionizable sites in proteins has been traditionally based on numerical solutions of the Poisson-Boltzmann equation carried out using a high-resolution protein structure. In this paper, we present a method based on continuous constant pH molecular dynamics (CPHMD) simulations, which allows the first-principles description of protein ionization equilibria. Our method utilizes an improved generalized Born implicit solvent model with an approximate Debye-Hückel screening function to account for salt effects and the replica-exchange (REX) protocol for enhanced conformational and protonation state sampling. The accuracy and robustness of the present method are demonstrated by 1 ns REX-CPHMD titration simulations of 10 proteins, which exhibit anomalously large pK(a) shifts for the carboxylate and histidine side chains.
Khandogin J* and Brooks CL III
2005Constant pH molecular dynamics simulation with proton tautomerism.
Biophys J 89: 141-157, 2005
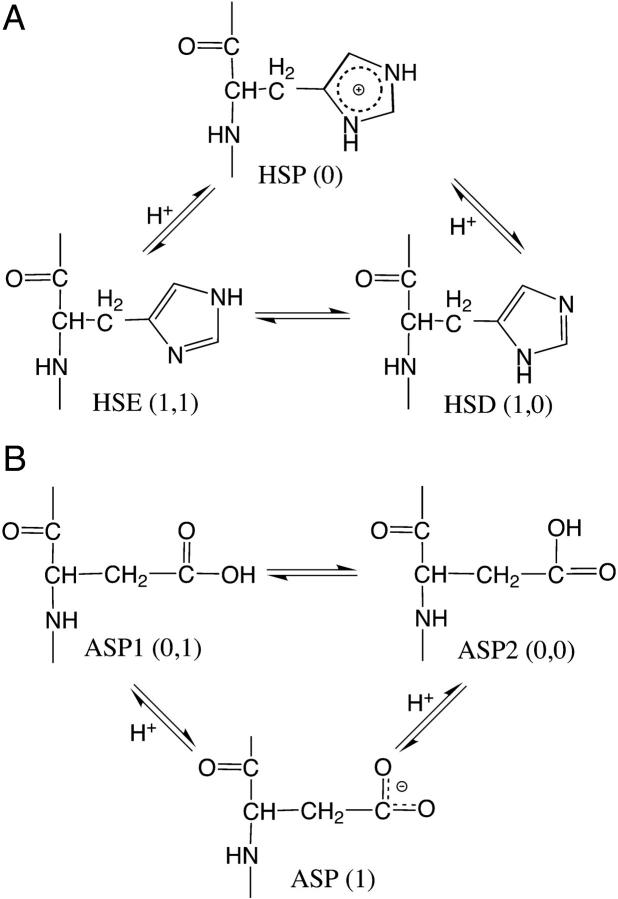
The current article describes a new two-dimensional lambda-dynamics method to include proton tautomerism in continuous constant pH molecular dynamics (CPHMD) simulations. The two-dimensional lambda-dynamics framework is used to devise a tautomeric state titration model for the CPHMD simulations involving carboxyl and histidine residues. Combined with the GBSW implicit solvent model, the new method is tested on titration simulations of blocked histidine and aspartic acid as well as two benchmark proteins, turkey ovomucoid third domain (OMTKY3) and ribonuclease A (RNase A). A detailed analysis of the errors inherent to the CPHMD methodology as well as those due to the underlying solvation model is given. The average absolute error for the computed pKa values in OMTKY3 is 1.0 pK unit. In RNase A the average absolute errors for the carboxyl and histidine residues are 1.6 and 0.6 pK units, respectively.