Read our papers in Zotero
Clayton J, Kozell LB, Eshleman AJ, Bloom SH, Schutzer WE, Abbas AI, Stavitskaya L*, Shen J*
Slow Dissociation of Nitazenes from the mu-Opioid Receptor Underlies the Challenge of Overdose Reversal
Submitted, 2026
Dayhoff II GW, Kortzak D, Liu R, Shen M, and Shen J*
Illuminating the Druggable Proteome with an AI Protein Profiling Platform
In revision, 2026
DOI: bioRxiv
Shen M, Dayhoff II GW, Shen J*
Protein Electrostatic Properties are Fine-Tuned Through Evolution
In revision, 2026
DOI: bioRxiv
Clayton J, Farmer G, Glenn J, Mistry SN, Lane JR, Shi L, Stavitskaya L, Canals M, Shen J*
Membrane Permeability Drives the Extreme Potency of Fentanyl
JACS Au, 2026
DOI: jacsau.5c01666
Case, Cerutti, ..., Shen, ..., Nagan*, Merz Jr.*
Recent Developments in Amber Biomolecular Simulations
J Chem Inf Model 65: 7835–7843, 2025
DOI: 10.1021/acs.jcim.5c01063
Romany A, Payne GF, Shen J*
How Electric Field Remodels the Nanofibril Structure of Chitosan Hydrogels: The Role of Dewetting during Electro-Assembly
ACS Appl Nano Mater 8: 15299–15308, 2025
DOI: 10.1021/acsanm.5c02691
Chen D, Liu G, Du H, Wee J, Wang R, Chen J, Shen J*, and Wei G-W*
Drug Resistance Predictions Based on a Directed Flag Transformer
Adv Sci, e02756, 2025
DOI: 10.1002/advs.202502756
Shen M, Kortzak D, Ambrozak S, Bhatnagar S, Buchanan I, Liu R, Shen J*
KaMLs for Predicting Protein pK a Values and Ionization States: Are Trees All You Need?
J Chem Theory Comput 21: 1446–1458, 2025
DOI: 10.1021/acs.jctc.4c01602
Clayton J, Shi L, Robertson MJ, Skiniotis G, Michaelides M, Stavitskaya L*, Shen J*
A Putative Binding Model of Nitazene Derivatives at the μ-Opioid Receptor
Neuropharmacology 273, 110437, 2025
DOI: S0028390825001431
Peeples CA, Liu R, and Shen J*
Force Field Limitations of All-Atom Continuous Constant pH Molecular Dynamics
J Phys Chem B 128: 11616–11624, 2024
PMCID: PMC11398383 DOI: 10.1021/acs.jpcb.4c05971
Hwang W*,..., Shen J,..., Brooks III CL*, Brooks BR*, and Karplus M
CHARMM at 45: Enhancements in accessibility, functionality, and speed
J Phys Chem B 128: 9979-10042, 2024
PMCID: PMC11492285 DOI: 10.1021/acs.jpcb.4c04100
Gogal R, Nessler A, Thiel A, Bernabe H, Grove RC, Cousineau L, Litman J, Miller J, Qi G, Speranza M, Tollefson M, Fenn T, Michaelson J, Okada O, Piquemal J-P, Ponder JW, Shen J, Smith R, Yang W, Ren R, and Schnieders M*
Force Field X: A Computational Microscope to Study Genetic Variation and Organic Crystals Using Theory and Experiment
J Chem Phys 161, 012501, 2024
DOI: 10.1063/5.0214652
Shen M, Huang Y*, Cai Z, Cherny, VV, DeCoursey TE, and Shen J*
Interior pH Sensing Residue of Human Voltage-Gated Proton Channel Hv1 is Histidine 168
Biophys J 123, 4211-4220, 2024
DOI: 10.1016/j.bpj.2024.07.027
Oh M, Shen M, Liu R, Stavitskaya L*, and Shen J*
Machine Learned Classification of Ligand Intrinsic Activity at Human μ-Opioid Receptor
ACS Chem Neurosci 15: 2842–2852, 2024
PMCID: PMC DOI: 10.1021/acschemneuro.4c00212 Paper Chat: Science Cast
Ibrahim M, Sun X, de Oliveira M, Liu R, Clayton R, El Kilani H, Shen J*, Hilgenfeld R*
Why is the Omicron main protease of SARS-CoV-2 less stable than its wild-type counterpart? A crystallographic, biophysical, and theoretical study of the free enzyme and its complex with inhibitor 13b-K
hLife 2: 419-433, 2024
DOI: 10.1016/j.hlife.2024.06.003 Paper Chat: Science Cast
Ma S*, Patel H, Peeples CA, Shen J*
QM/MM Simulations of Afatinib-EGFR Addition: the Role of β-Dimethylaminomethyl Substitution
J Chem Theory Comput 20: 5528–5538, 2024
DOI: 10.1021/acs.jctc.4c00290 Paper Chat: Science Cast
Clayton J, Romany A, Matenoglou E, Gathiotis E, Poulikakos PI, and Shen J*
Mechanism of Dimer Selectivity and Binding Cooperativity of BRAF inhibitors
eLife, 2024
PMCID: PMC10760002 DOI: 10.7554/eLife.95334.2 Paper Chat: Science Cast
Thiel A, Speranza MJ, Jadhav S, Stevens LL, Unruh DK, Ren P, Ponder JW, Shen J, Schnieders M*
Constant-pH Simulations with the Polarizable Atomic Multipole AMOEBA Force Field
J Chem Theory Comput 20: 2921-2933, 2024
PMCID: PMC3806652 DOI: 10.1021/acs.jctc.3c01180
Liu R, Clayton J, Shen M, Bhatnagar S, and Shen J*
Machine Learning Models to Interrogate Proteome-wide Covalent Ligandabilities Directed at Cysteines
JACS Au 4: 1374–1384, 2024
PMCID: PMC10473668 DOI: 10.1021/jacsau.3c00749 Paper Chat: Science Cast
Romany A, Payne GF, Shen J*
Effect of Acetylation on the Nanofibril Formation of Chitosan From All-Atom de novo Self-Assembly Simulations
Molecules 29: 561, 2024
DOI: 10.3390/molecules29030561
Romany A, Payne GF, Shen J*
Mechanism of the Temperature-Dependent Self-Assembly and Polymorphism of Chitin
Chem Mater 35: 6472–6481, 2023
DOI: 10.1021/acs.chemmater.3c01313
Liu R, Vazquez-Montelongo EA, Ma S*, and Shen J*
Quantum Descriptors for Predicting and Understanding the Structure-Activity Relationships of Covalent Warheads
J Chem Inf Model 63: 4912-4923, 2023
DOI: 10.1021/acs.jcim.3c00720
Liu Y, Kim E, Lei M, Wu S, Yan K, Shen J, Bentley W, Qu X, Shi X, Payne, GF*
Electro-bio-fabrication: Coupling Electrochemical and Biomolecular Methods to Create Functional Bio-based Hydrogels
Biomacromolecules 24: 2409-2432, 2023
DOI: 10.1021/acs.biomac.3c00132
Romany E, Liu R, Zhan S, Clayton J, and Shen J*
Analysis of the ERK Pathway Cysteinome for Targeted Covalent Inhibition of RAF and MEK Kinases
J Chem Inf Model 63: 2483–2494, 2023
PMCID: PMCID DOI: 10.1021/acs.jcim.3c00014
Mahinthichaichan P, Liu R, Vo QN, Ellis CR, Stavitskaya L*, and Shen J*
Structure-Kinetics Relationships of Opioids from Molecular Dynamics Simulations and Machine Learning
J Chem Inf Model 63: 2196–2206, 2023
PMCID: PMC10028827 DOI: 10.1021/acs.jcim.3c00069 Paper Chat: Science Cast
Clayton J, de Oliveira V M, Ibrahim M F, Sun XY, Mahinthichaichan P, Shen M, Hilgenfeld R, Shen J*.
Integrative Approach to Dissect the Drug Resistance Mechanism of the H172Y Mutation of SARS-CoV-2 Main Protease
J Chem Inf Mode 63: 3521–3533, 2023
PMCID: PMC9387124 DOI: 10.1021/acs.jcim.3c00344
David A Case, H Metin Aktulga, Kellon Belfon, IY Ben-Shalom, Scott R Brozell, DS Cerutti, TE Cheatham III, GA Cisneros, VWD Cruzeiro, TA Darden, RE Duke, G Giambasu, MK Gilson, H Gohlke, AW Goetz, R Harris, S Izadi, SA Izmailov, C Jin, K Kasavajhala, MC Kaymak, E King, A Kovalenko, T Kurtzman, TS Lee, S LeGrand, P Li, C Lin, J Liu, T Luchko, R Luo, M Machado, V Man, M Manathunga, KM Merz, Y Miao, O Mikhailovskii, G Monard, H Nguyen, KA O’Hearn, A Onufriev, F Pan, S Pantano, R Qi, A Rahnamoun, DR Roe, A Roitberg, C Sagui, S Schott-Verdugo, J Shen, CL Simmerling, NR Skrynnikov, J Smith, J Swails, RC Walker, J Wang, H Wei, RM Wolf, X Wu, Y Xue, DM York, S Zhao, PA Kollman
Amber22
Software release, 2022
de Oliveira VM, Liu R, Shen J*
Constant pH Molecular Dynamics Simulations: Current Status and Recent Applications
Curr Opin Struct Biol 77: 102498, 2022
PMCID: PMC9933785 DOI: 10.1016/j.sbi.2022.102498
Yang R, Han S, Clayton C, Haghighatian M, Tsai C-C, Yao Y, Li P, Shen J, Zhou Q*
A proof-of-concept inhibitor of endothelial lipase suppresses triple-negative breast cancer cells by hijacking the mitochondrial function
Cancers 14: 3763, 2022
PMCID: PMC9367514 DOI: 10.3390/cancers14153763
Harris, JA, Liu, R, Martins de Oliveira, V, Vazquez Montelongo, E, Henderson, JA, and Shen, J
GPU-Accelerated All-atom Particle-Mesh Ewald Continuous Constant pH Molecular Dynamics in Amber
J Chem Theory Comput 18: 7510–7527, 2022
PMCID: PMC10130738 DOI: 10.1021/acs.jctc.2c00586
Liu R, Henderson JA, Harris JA, Huang Y, de Oliveira MV, and Shen J*
A Guide to the Continuous Constant pH Molecular Dynamics Methods in Amber and CHARMM [v1.0]
Liv J Comput Mol Sci 4: 1563, 2022
PMCID: PMC9910290 DOI: 10.33011/livecoms.4.1.1563
Henderson JA and Shen J*
Exploring the pH- and Ligand-Dependent Flap Dynamics of Malarial Plasmepsin II.
J Chem Inf Model 62: 150–158, 2022
PMCID: PMC8812262 DOI: 10.1021/acs.jcim.1c01180
Liu R, Verma N, Henderson JA, Zhan S, and Shen J*
Profiling Cysteines in MAP Kinases for Targeted Covalent Inhibitor Design.
RSC Med Chem 13: 54-63, 2022
PMCID: PMC8792824 DOI: 10.1039/D1MD00277E
Liu R, Zhan S, Che Y, Shen J*
Reactivities of the Front Pocket Ncap Cysteines in Human Kinases.
J Med Chem 65: 1525-1535, 2022
PMCID: PMC8812259 DOI: 10.1101/2021.06.28.450170
Mahinthichaichan P, Vo Q, Ellis CR, and Shen J*
Kinetics and Mechanism of Fentanyl Dissociation from the mu-Opioid Receptor.
JACS Au 1: 2208–2215, 2021
PMCID: PMC8715493 DOI: 10.1021/jacsau.1c00341
Vo QN, Mahinthichaichan P, Shen J*, and Ellis CR*
How μ-opioid receptor recognizes fentanyl.
Nat Commun 12: 984, 2021
PMCID: PMC7881245 DOI: 10.1038/s41467-021-21262-9
Huang YD, Henderson JA, and Shen J*
Continuous constant pH molecular dynamics of transmembrane proteins.
Methods Mol Biol 2302: 275–287, 2021
PMCID: PMC8062021 DOI: 10.1101/2020.08.06.239772
Ma S*, Henderson JA, and Shen J*
Exploring the pH-dependent structure-dynamics-function relationship of human renin.
J Chem Inf Model 61: 400-407, 2021
PMCID: PMC7855609 DOI: 10.1021/acs.jcim.0c01201
Verma N, Henderson JA, and Shen J*
Proton-Coupled Conformational Activation of SARS Coronavirus Main Proteases and Opportunity for Designing Small-Molecule Broad-Spectrum Targeted Covalent Inhibitors.
J Am Chem Soc 142: 21883–21890, 2020
PMCID: PMC7754784 DOI: 10.1021/jacs.0c10770
Henderson JA, Verma N, Harris R, Liu R, and Shen J*
Assessment of proton-coupled conformational dynamics of SARS and MERS coronavirus papain-like proteases: Implication for designing broad-spectrum antiviral inhibitors.
J Chem Phys 153: 115101, 2020
PMCID: PMC7337382 DOI: 10.1063/5.0020458
Harris RC, Liu R, and Shen J*
Predicting reactive cysteines with implicit-solvent based continuous constant pH molecular dynamics in Amber.
J Chem Theory Comput 16: 3689-3698, 2020
PMCID: PMC7772776 DOI: 10.1021/acs.jctc.0c00258
Henderson JA, Beckstein O, and Shen J*
Alternative proton-binding site and long-distance coupling in Escherichia coli sodium–proton antiporter NhaA.
Proc Natl Acad Sci USA, 117: 25517-25522, 2020
PMCID: PMC7568328 DOI: 10.1073/pnas.2005467117
Wu S, Shi X, Yan K, Li J, Huynh R, Raub C, Shen J and Payne GF*
Electrical cuing of chitosan's mesoscale organization.
React Funct Polym 148: 104492, 2020
PMCID: None DOI: 10.1016/j.reactfunctpolym.2020.104492
Mahinthichaichan P, Tsai CC, Payne, GF, and Shen J*
Polyelectrolytes in electric field: disparate conformational behavior along an amino-polysaccharide chain.
ACS Omega 5: 12016-12026, 2020
PMCID: PMC7271050 DOI: 10.1021/acsomega.0c00164
David A Case, H Metin Aktulga, Kellon Belfon, IY Ben-Shalom, Scott R Brozell, DS Cerutti, TE Cheatham III, GA Cisneros, VWD Cruzeiro, TA Darden, RE Duke, G Giambasu, MK Gilson, H Gohlke, AW Goetz, R Harris, S Izadi, SA Izmailov, C Jin, K Kasavajhala, MC Kaymak, E King, A Kovalenko, T Kurtzman, TS Lee, S LeGrand, P Li, C Lin, J Liu, T Luchko, R Luo, M Machado, V Man, M Manathunga, KM Merz, Y Miao, O Mikhailovskii, G Monard, H Nguyen, KA O’Hearn, A Onufriev, F Pan, S Pantano, R Qi, A Rahnamoun, DR Roe, A Roitberg, C Sagui, S Schott-Verdugo, J Shen, CL Simmerling, NR Skrynnikov, J Smith, J Swails, RC Walker, J Wang, H Wei, RM Wolf, X Wu, Y Xue, DM York, S Zhao, PA Kollman
Amber20
Software release, 2020
Harris RC and Shen J*
GPU-accelerated implementation of continuous constant pH molecular dynamics in Amber: pKa predictions with single pH simulations.
J Chem Inf Model 59: 4821-4832, 2019
PMCID: PMC6934042 DOI: 10.1021/acs.jcim.9b00754
Tsai CC, Yue Z, and Shen J*
How electrostatic coupling enables conformational plasticity in a tyrosine kinase.
J Am Chem Soc 141: 15092-15101, 2019
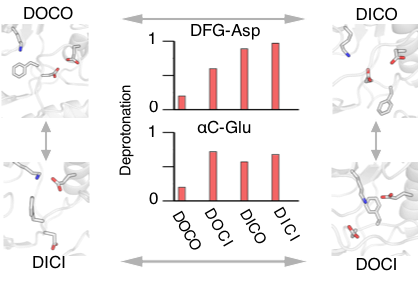
The ability to predictively model various conformational states could accelerate selectivity inhibitor design. We present, to our best knowledge, the first predictive computational study that captured the four major types of kinase conformations starting from a single crystal structure without employing a target structure, mutation, or bias. Our data revealed, for the first time, how protonation state changes of key residues facilitate the conformational transitions, providing a direct support and explanation for a long-standing hypothesis regarding the protonation-dependent DFG flip. Our simulations also uncovered intermediate states, among which a unique inactive state, which can be tested in selective inhibitor design.
Kim E, Li J, Kang M, Kelly DL, Chen S, Napolitano A, Panzella L, Shi X, Yan K, Wu S, Shen J, Bentley, WE, and Payne GF*
Redox is a global biodevice information processing modality.
Proc IEEE: Special Issue on "Molecular Communications and Networking" 107: 1402-1424, 2019
PMCID: PMC7036710 DOI: 10.1109/JPROC.2019.2908582
Liu R, Yue Z, Tsai CC, and Shen J*
Assessing lysine and cysteine reactivities for designing targeted covalent kinase inhibitors.
J Am Chem Soc 141: 6553-6560, 2019
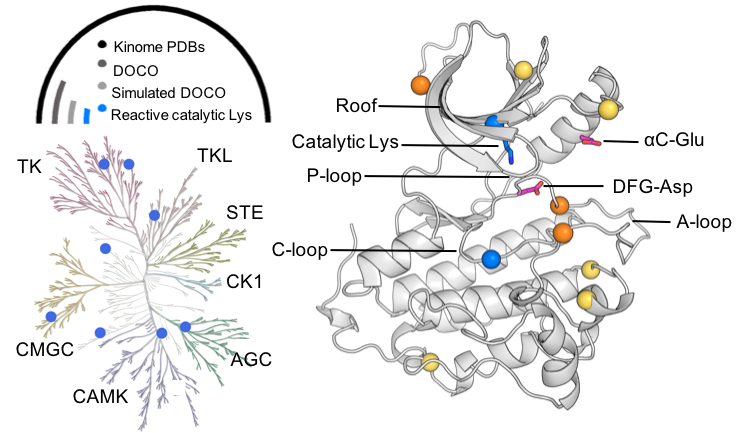
We present a computational tool to assess the nucleophilicities of lysines and cysteines to accelerate covalent drug design. The tool enabled us to discover that kinases in a rare inactive conformation can be covalently targeted at catalytic lysines. A new strategy for designing highly selective, lysine-targeted covalent kinase inhibitors was proposed to address the emergent drug resistance issue facing the cysteine-targeted strategy and help usher in the next generation covalent kinase inhibitor design.
Li J, Wu S, Kim E, Yan K, Liu H, Liu C, Dong H, Qu X, Shi X, Shen J, Bentley, WE, and Payne GF*
Electrobiofabrication: electrically-based fabrication with biologically-derived materials.
Biofabrication 11: 032002, 2019
PMCID: PMC7025432 DOI: 10.1088/1758-5090/ab06ea
Tsai CC, Payne GF, and Shen J
Exploring pH-responsive, switchable crosslinking mechanisms for programming reconfigurable hydrogels based on aminopolysaccharides.
Chem Mater 30: 8597-8605, 2018
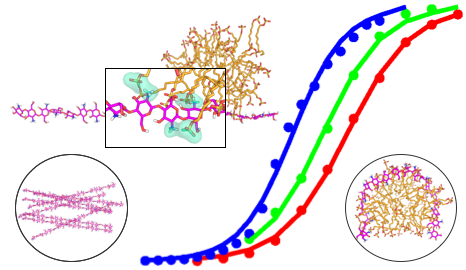
Designing dynamic, stimuli-responsive, reconfigurable materials has growing interest in academia and industry. We report a computational study that provided novel, atomically-detailed physical insights for a pH-responsive, reconfigurable system. pKa gradients are known to be integral to protein structures and functions; our work presents the first theoretical demonstration of a pKa gradient for a highly dynamical polysaccharide system and how it allows a persistent but erasable gradient in the structural and mechanical properties of the hydrogel.
Henderson JA, Harris RC, Tsai CC, and Shen J*
How ligand protonation state controls water in protein-ligand binding.
J Phys Chem Lett 9: 5440-5444, 2018
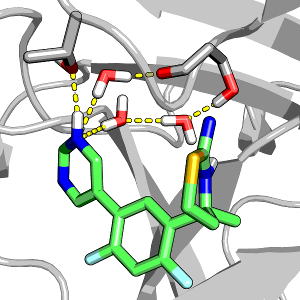
The role of water in protein-ligand has been intensely studied in recent years; however, how ligand protonation state change perturbs water has not been investigated. Here we use two homologues aspartyl proteases to illustrate how modeling protein-ligand binding while allowing ligand titration can further advance our understanding of the role of water in modulating the inhibitor selectivity for a particular target.
Shen J*
Zooming in on a multi-drug transporter reveals details of asymmetric protonation.
Proc Natl Acad Sci USA 115: 6080-6082, 2018
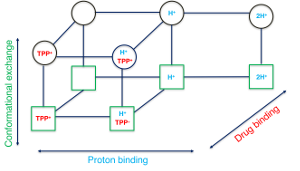
Nature is efficient; why would it require two protons if one is enough to do the job? This commentary article discusses a recent computational work in the context of our current understanding of how proton translocation mediates the active transport of drugs and substrates across the biological membrane.
D.A. Case, I.Y. Ben-Shalom, S.R. Brozell, D.S. Cerutti, T.E. Cheatham, III, V.W.D. Cruzeiro, T.A. Darden, R.E. Duke, D. Ghoreishi, M.K. Gilson, H. Gohlke, A.W. Goetz, D. Greene, R Harris, N. Homeyer, S. Izadi, A. Kovalenko, T. Kurtzman, T.S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D.J. Mermelstein, K.M. Merz, Y. Miao, G. Monard, C. Nguyen, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R. Qi, D.R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C.L. Simmerling, J. Smith, R. Salomon-Ferrer, J. Swails, R.C. Walker, J. Wang, H. Wei, R.M. Wolf, X. Wu, L. Xiao, D.M. York, and P.A. Kollman
AMBER18
Software release, 2018
Huang YD, Harris RC, Shen J*
Generalized Born based continuous constant pH molecular dynamics in Amber: implementation, benchmarking, and analysis.
J Chem Inf Model 58:1372-1383, 2018

We report the CPU implementation of the CpHMD method based on the GBNeck2 generalized Born (GB) implicit-solvent model in the pmemd engine of the Amber molecular dynamics package. Benchmark data based on 11 proteins are discussed. The related software is disseminated through Amber18.
Huang YD, Yue Z, Tsai CC, Henderson JA, and Shen J*
Predicting catalytic proton donors and nucleophiles in enzymes: how adding dynamics helps elucidate the structure-function relationships.
J Phys Chem Lett 9: 1179-1184, 2018
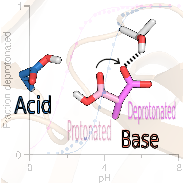
Despite the importance, prediction of catalytic proton donors and nucleophiles has remained an unsolved problem. Here we tested three types of state-of-the-art computational methods to predict the pKa’s of buried and coupled carboxyl dyads in enzymes. We asked the question what makes a residue proton donor vs. nucleophile. Our study revealed a surprisingly simple trend, which is not apparent from crystal structures, empirical or traditional pKa calculations.
Yue Z and Shen J*
pH-dependent cooperativity and existence of a dry molten globule in the folding of a mini-protein BBL.
Phys Chem Chem Phys, 20, 3523-3530, 2018
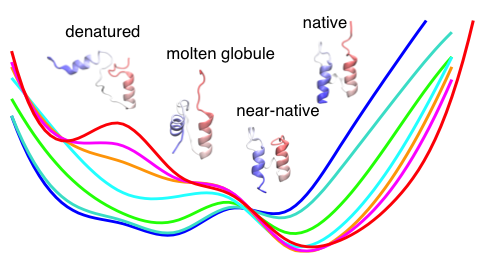
pH plays an important role in protein dynamics, stability, and folding; however, detailed mechanisms remain poorly understood. We carried out the first explicit-solvent protein folding simulation in a pH-dependent manner. Remarkably, the data revealed an unfolding intermediate bearing the signature of a dry molten globule, which is currently hypothesized as a universal intermediate in protein folding.
Yan K, Liu Y, Zhang J, Correa SO, Shang W, Tsai CC, Bentley WE, Shen J, Scarcelli G, Raub CB, Shi XW, Payne GF*
Electrical programming of soft matter: using temporally varying electrical inputs to spatially control self assembly.
Biomacromolecules 19: 364-373, 2018
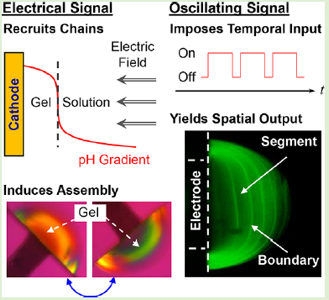
Top-down fabrication methods lack the capabilities to generate complex matrix architectures observed in biology. Here we show that temporally varying electric signals can cue a self-assembling polysaccharide to controllably form a hydrogel with complex internal patterns.
Yue Z, Chen W, Zgurskaya, H, and Shen J*
Constant pH molecular dynamics reveals how proton release drives the conformational transition of a transmembrane efflux pump.
J Chem Theory Comput 13: 6405-6414, 2017
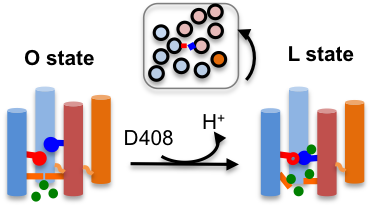
AcrB is the inner-membrane transporter of the AcrAB-TolC efflux complex in E. coli which confers resistance to antibiotics. Crystal structures have revealed three distinct states in the drug transport cycle; however, detailed mechanism remains unknown. Our study offered convincing support for the 1:1 drug:proton stoichiometry by depicting how proton release triggers the conformational transition, in agreement with the crystal structure.
Harris RC, Tsai CC, Ellis CR, and Shen J*
Proton-coupled conformational allostery modulates the inhibitor selectivity for β-secretase.
J Phys Chem Lett 8: 4832-4837, 2017
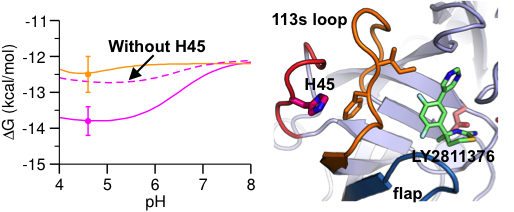
Many pharmaceutical targets, such as aspartyl proteases and kinases, exhibit pH-dependent dynamics and functions. Accurate prediction of their binding free energies is challenging, and current computational studies neglect the effects of pH and changes in protonation states. We demonstrated a new protocol to improve the predictive power of current binding free energy calculations. The allosteric mechanism discovered here could lead to exciting opportunities in drug design.
Tsai CC, Morrow BH, Chen W, Payne GP, and Shen J*
Towards understanding the environmental control of hydrogel film properties: how salt modulates the flexibility of chitosan chains.
Macromolecules 50: 5946-5952, 2017
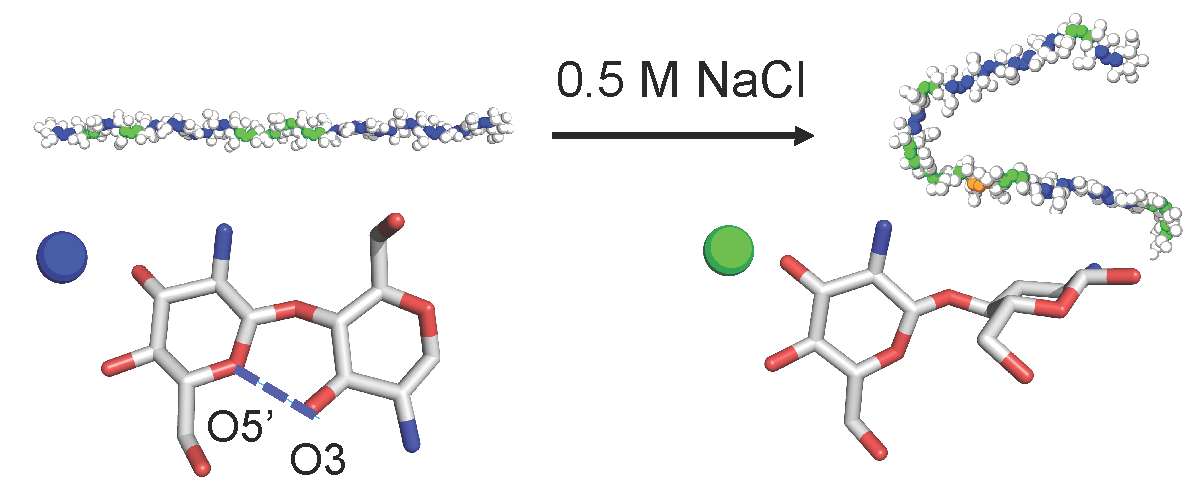
Aminopolysaccharides can form complex structures with the properties highly dependent on environmental conditions; however, molecular details are not understood. We found that, contrary to common belief, chitosan chains have a high intrinsic flexibility and salt increases the flexibility by shifting the population from extended to bent glycosidic backbone conformation.
Deredge DJ, Huang W, Huib C, Matsumura H, Yue Z, Moenne-Loccoz P, Shen J, Wintrode PL, and Wilks A*
Ligand-induced allostery in the interaction of the Pseudomonas aeruginosa heme binding protein with heme oxygenase.
Proc Natl Acad Sci USA 114:3421-3426, 2017
PMCID: PMC5380046 DOI: 10.1073/pnas.1606931114
Ellis CR, Tsai CC, Lin FY, and Shen J*
Conformational dynamics of cathepsin D and binding to a small-molecule BACE1 inhibitor.
J Comput Chem 38:1260-1269, 2017
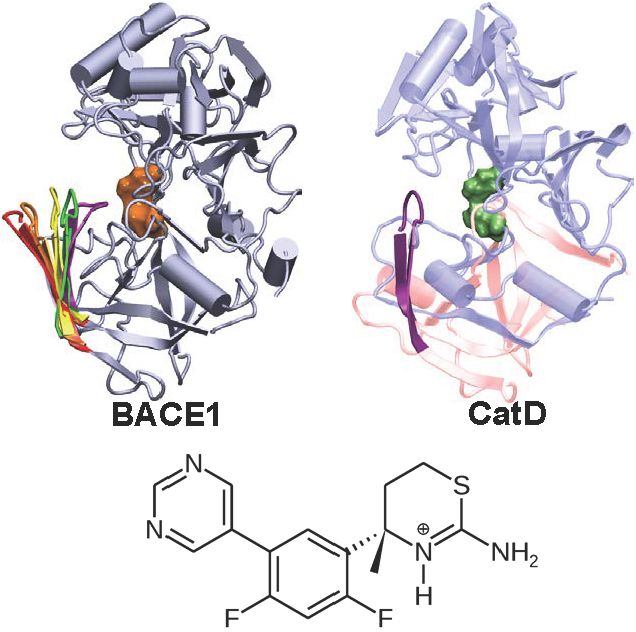
Targeting the aspartyl protease BACE1 with small-molecule inhibitors offers a promising route for treatment of Alzheimer's disease. However, developing inhibitors that can selectively target BACE1 in favor of other proteases, especially cathepsin D, has presented significant challenges. We reported the first computational study that characterizes the conformational dynamics of capthesin D and how it binds a BACE1 small-molecule inhibitor.
Chen W, Huang YD and Shen J*
Conformational activation of a transmembrane proton channel from constant pH molecular dynamics.
J Phys Chem Lett 7: 3961-3966, 2016
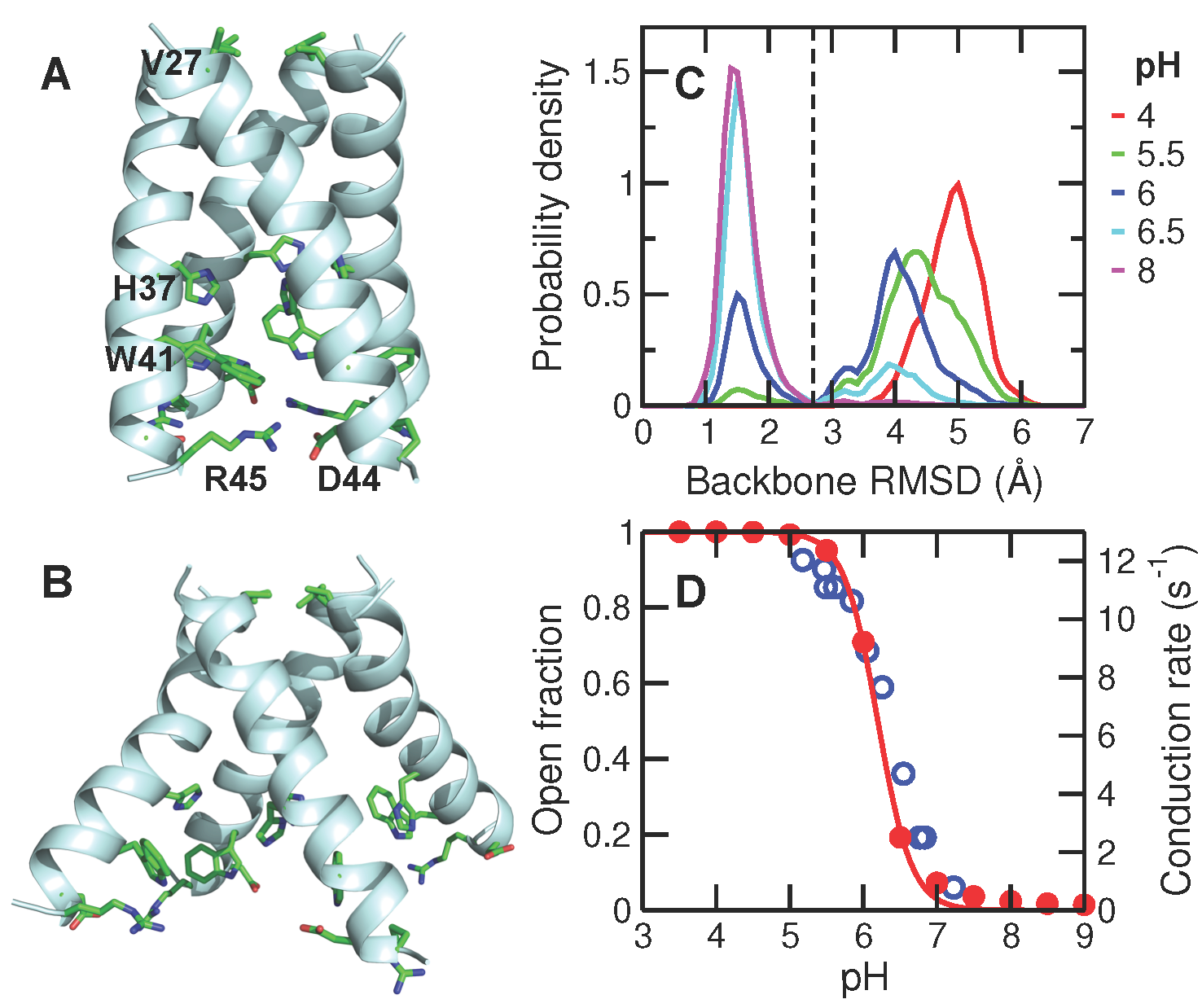
Proton-coupled transmembrane proteins play important roles in human health and diseases. We performed the first computer simulation that 1) directly describes the proton-coupled conformational activation of a transmembrane channel with fully atomic detail; 2) accurately determines the stepwise acid-base constants of a transmembrane channel; 3) provides the proton-coupled free energy of channel activation. The presented methodologies and major findings are generalizable for studies of proton-coupled channels and transporters.
Huang YD, Chen W, and Shen J*
Continuous constant pH molecular dynamics with particle-mesh Ewald and titratable water.
J Chem Theory Comput 12: 5411-5421, 2016
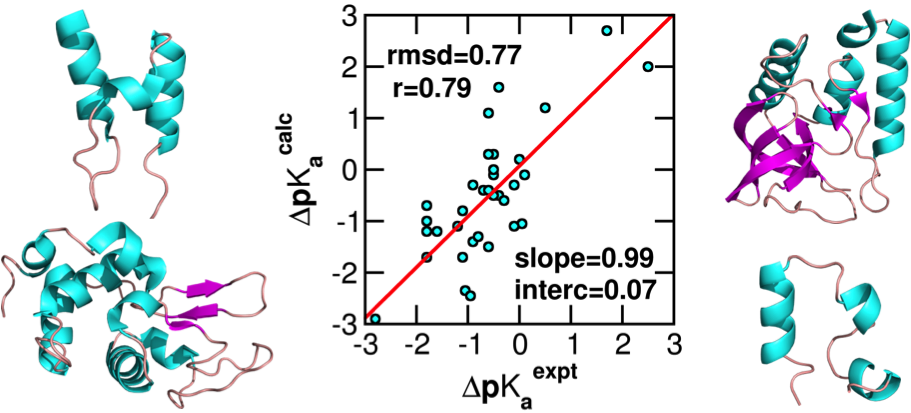
Development of a pH stat to control solution pH in biomolecular simulations remains a outstanding goal. In recent years, the constant pH molecular dynamics methods have emerged; however, the accuracy and generality have been hampered by the use of implicit-solvent models or truncation-based electrostatic schemes. We reported the first implementation of the all-atom PME-based continuous constant pH method and benchmark results on a set of commonly used proteins.
Huang YD, Chen W, Dotson DL, Beckstein O, and Shen J*
Mechanism of pH-dependent activation of the sodium-proton antiporter NhaA.
Nat Commun 7: 12940, 2016
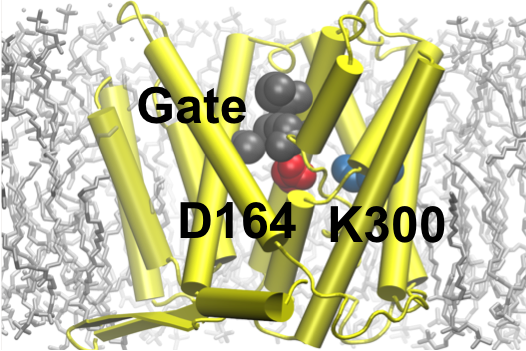
NhaA is the principal sodium-proton antiporter in E. Coli., essential for cellular sodium and pH homeostasis. Abundant experimental and computational studies as well as two high-resolution crystal structures have been published; however, the pH-regulated mechanism at the molecular-level remains elusive. We reported the first computational study that offers an atomically-detailed view of the pH-dependent activation and key conformational events in the sodium-proton exchange cycle of NhaA.
Kim E, Liu Y, Ben-Yoav H, Winkler TE, Yan K, Shi S, Shen J, Kelly DL, Ghodssi R, Bentley WE, and Payne GF*
Fusing sensor paradigms to acquire chemical information: an integrative role for smart biopolymeric hydrogels.
Adv Healthcare Mater 5: 2595-2616, 2016
PMCID: PMC5485850 DOI: 10.1002/adhm.201600516
Ellis CR, Tsai CC, Hou XJ, and Shen J*
Constant pH molecular dynamics reveals pH-modulated binding of two small-molecule BACE1 inhibitors.
J Phys Chem Lett 7: 944-949, 2016
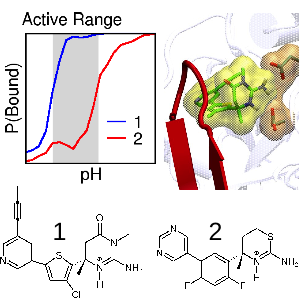
Targeting the aspartyl protease BACE1 with small-molecule inhibitors offers a promising route for treatment of Alzheimer's disease. However, the intricate pH dependence of BACE1 function and inhibitor efficacy has posed major challenges. We reported the first simulation that uncovers the atomic details of pH-dependent binding of two drug candidates for BACE1. Our work demonstrated the drastic contrast in affinity of the two inhibitors is due to the subtle differences in the protonation behavior of the protein-inhibitor complex.